Electrochemical Determination of Hydroxyurea in a Complex Biological Matrix Using MoS2-Modified Electrodes and Chemometrics

 ,
,  , ,
, , 
Abstract
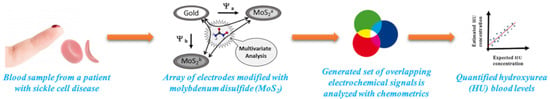
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Consumables and Equipment
2.2. Buffered, Simulated Serum, and Serum Sample Preparation
2.3. Electrodeposition of MoS2
2.4. Electrochemical Characterization
2.5. Electrochemical Sensing of Hydroxyurea, Uric Acid, and L-Ascorbic Acid in PBS and Simulated Serum
2.6. Chemometric Analysis
3. Results and Discussion
3.1. Electrodeposition of MoS2 on Polycrystalline Gold Electrodes
3.2. Electrochemical Signatures of Hydroxyurea, Uric Acid, and Ascorbic Acid
3.3. Analysis of Hydroxyurea in a Simulated Serum Using Chemometrics
3.4. Analysis of Hydroxyurea in Real Human Serum with an Electrode Array
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- 17th Expert Committee on the Selection and Use of Essential Medicines. 2010. Available online: https://www.who.int/selection_medicines/committees/expert/17/en/ (accessed on 23 December 2020).
- de Montalembert, M.; Bachir, D.; Hulin, A.; Gimeno, L.; Mogenet, A.; Bresson, J.L.; Macquin-Mavier, I.; Roudot-Thoraval, F.; Astier, A.; Galactéros, F. Pharmacokinetics of hydroxyurea 1000 mg coated breakable tablets and 500 mg capsules in pediatric and adult patients with sickle cell disease. Haematologica 2006, 91, 1685–1688. [Google Scholar] [PubMed]
- Ware, R.E.; Despotovic, J.M.; Mortier, N.A.; Flanagan, J.M.; He, J.; Smeltzer, M.P.; Kimble, A.C.; Aygun, B.; Wu, S.; Howard, T.; et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011, 118, 4985–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGann, P.T.; Niss, O.; Dong, M.; Marahatta, A.; Howard, T.A.; Mizuno, T.; Lane, A.; Kalfa, T.A.; Malik, P.; Quinn, C.T.; et al. Robust clinical and laboratory response to hydroxyurea using pharmacokinetically guided dosing for young children with sickle cell anemia. Am. J. Hematol. 2019, 94, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; McGann, P.T.; Mizuno, T.; Ware, R.E.; Vinks, A.A. Development of a pharmacokinetic-guided dose individualization strategy for hydroxyurea treatment in children with sickle cell anemia. Br. J. Clin. Pharmacol. 2016, 81, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Marahatta, A.; Ware, R.E. Hydroxyurea: Analytical techniques and quantitative analysis. Blood Cells Mol. Dis. 2017, 67, 135–142. [Google Scholar] [CrossRef]
- Marahatta, A.; Dong, M.; Opoka, R.; McElhinney, K.E.; Latham, T.S.; John, C.C.; Vinks, A.A.; Ware, R.E. Pharmacokinetics of hydroxyurea therapy in African children with sickle cell anemia: Noharm ancillary PK study. Blood 2017, 130, 2252. [Google Scholar]
- Vinks, A.A.; Peck, R.W.; Neely, M.; Mould, D.R. Development and implementation of electronic health record–integrated model-informed clinical decision support tools for the precision dosing of drugs. Clin. Pharmacol. Ther. 2020, 107, 129–135. [Google Scholar] [CrossRef]
- Smart, L.R.; Hernandez, A.G.; Ware, R.E. Sickle cell disease: Translating clinical care to low-resource countries through international research collaborations. Semin. Hematol. 2018, 55, 102–112. [Google Scholar] [CrossRef]
- McGann, P.T.; Hernandez, A.G.; Ware, R.E. Sickle cell anemia in sub-Saharan Africa: Advancing the clinical paradigm through partnerships and research. Blood 2017, 129, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Hai, X.; Guo, M.; Gao, C.; Zhou, J. Quantification of hydroxyurea in human plasma by HPLC–MS/MS and its application to pharmacokinetics in patients with chronic myeloid leukaemia. J. Pharm. Biomed. Anal. 2017, 137, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Pujari, M.P.; Barrientos, A.; Muggia, F.M.; Koda, R.T. Determination of hydroxyurea in plasma and peritoneal fluid by high-performance liquid chromatography using electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1997, 694, 185–191. [Google Scholar] [CrossRef]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheuquepán, W.; Orts, J.M.; Rodes, A. Hydroxyurea electrooxidation at gold electrodes. In situ infrared spectroelectrochemical and DFT characterization of adsorbed intermediates. Electrochim. Acta 2017, 246, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Naik, K.M.; Alagur, M.M.; Nandibewoor, S.T. Electrochemical response of hydroxyurea by different voltammetric techniques at carbon paste electrode. Anal. Methods 2013, 5, 6947–6953. [Google Scholar] [CrossRef]
- Pathak, P.K.; Kumar, A.; Prasad, B.B. Functionalized nitrogen doped graphene quantum dots and bimetallic Au/Ag core-shell decorated imprinted polymer for electrochemical sensing of anticancerous hydroxyurea. Biosens. Bioelectron. 2019, 127, 10–18. [Google Scholar] [CrossRef]
- Naik, K.M.; Ashi, C.R.; Nandibewoor, S.T. Anodic voltammetric behavior of hydroxyurea and its electroanalytical determination in pharmaceutical dosage form and urine. J. Electroanal. Chem. 2015, 755, 109–114. [Google Scholar] [CrossRef]
- Sun, H.; Chao, J.; Zuo, X.; Su, S.; Liu, X.; Yuwen, L.; Fan, C.; Wang, L. Gold nanoparticle-decorated MoS2 nanosheets for simultaneous detection of ascorbic acid, dopamine and uric acid. RSC Adv. 2014, 4, 27625–27629. [Google Scholar] [CrossRef]
- Legrand, T.; Rakotoson, M.-G.; Galactéros, F.; Bartolucci, P.; Hulin, A. Determination of hydroxyurea in human plasma by HPLC-UV using derivatization with xanthydrol. J. Chromatogr. B 2017, 1064, 85–91. [Google Scholar] [CrossRef]
- Naik, K.M.; Nandibewoor, S.T. Novel electroanalysis of hydroxyurea at glassy carbon and gold electrode surfaces. J. Electrochem. Sci. Eng. 2014, 4, 111–121. [Google Scholar] [CrossRef]
- Winquist, F. Voltammetric electronic tongues—Basic principles and applications. Microchim. Acta 2008, 163, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Holmin, S.; Spångeus, P.; Krantz-Rülcker, C.; Winquist, F. Compression of electronic tongue data based on voltammetry—A comparative study. Sens. Actuators B Chem. 2001, 76, 455–464. [Google Scholar] [CrossRef]
- Tarley, C.R.; Silveira, G.; dos Santos, W.N.; Matos, G.D.; da Silva, E.G.; Bezerra, M.A.; Miró, M.; Ferreira, S.L. Chemometric tools in electroanalytical chemistry: Methods for optimization based on factorial design and response surface methodology. Microchem. J. 2009, 92, 58–67. [Google Scholar] [CrossRef]
- Krzywinski, M.; Altman, N. Multiple linear regression. Nat. Methods 2015, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Velický, M.; Toth, P.S. From two-dimensional materials to their heterostructures: An electrochemist’s perspective. Appl. Mater. Today 2017, 8, 68–103. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.; Tan, B.; Huang, Y.; Zhao, H.; Dang, X.; Chen, J.; Jain, R. MoS2 nanostructures for electrochemical sensing of multidisciplinary targets: A review. TrAC 2018, 102, 75–90. [Google Scholar] [CrossRef]
- Venkata Subbaiah, Y.P.; Saji, K.J.; Tiwari, A. Atomically thin MoS2: A versatile nongraphene 2D material. Adv. Funct. Mater. 2016, 26, 2046–2069. [Google Scholar] [CrossRef]
- Zuo, P.; Jiang, L.; Li, X.; Li, B.; Xu, Y.; Shi, X.; Ran, P.; Ma, T.; Li, D.; Qu, L.; et al. Shape-controllable gold nanoparticle-MoS2 hybrids prepared by tuning edge-active sites and surface structures of MoS2 via temporally shaped femtosecond pulses. ACS Appl. Mater. Interfaces 2017, 9, 7447–7455. [Google Scholar] [CrossRef]
- Hai, X.; Zhou, W.; Wang, S.; Pang, H.; Chang, K.; Ichihara, F.; Ye, J. Rational design of freestanding MoS2 monolayers for hydrogen evolution reaction. Nano Energy 2017, 39, 409–417. [Google Scholar] [CrossRef]
- Ammam, M. Electrophoretic deposition under modulated electric fields: A review. RSC Adv. 2012, 2, 7633–7646. [Google Scholar] [CrossRef]
- Chávez-Valdez, A.; Boccaccini, A.R. Innovations in electrophoretic deposition: Alternating current and pulsed direct current methods. Electrochim. Acta 2012, 65, 70–89. [Google Scholar] [CrossRef]
- Gerein, N.J.; Haber, J.A. Effect of AC electrodeposition conditions on the growth of high aspect ratio copper nanowires in porous aluminum oxide templates. J. Phys. Chem. B 2005, 109, 17372–17385. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Li, H.; Park, S.; Park, J.; Han, H.S.; Nørskov, J.K.; Zheng, X.; Abild-Pedersen, F. Electrochemical generation of sulfur vacancies in the basal plane of MoS2 for hydrogen evolution. Nat. Commun. 2017, 8, 15113. [Google Scholar] [CrossRef] [PubMed]
- Escandar, G.M.; Damiani, P.C.; Goicoechea, H.C.; Olivieri, A.C. A review of multivariate calibration methods applied to biomedical analysis. Microchem. J. 2006, 82, 29–42. [Google Scholar] [CrossRef]
- Power-Hays, A.; Ware, R.E. Effective use of hydroxyurea for sickle cell anemia in low-resource countries. Curr. Opin. Hematol. 2020, 27, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Oleneva, E.; Khaydukova, M.; Ashina, J.; Yaroshenko, I.; Jahatspanian, I.; Legin, A.; Kirsanov, D. A simple procedure to assess limit of detection for multisensor systems. Sensors 2019, 19, 1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostra, M.; Ubide, C.; Vidal, M.; Zuriarrain, J. Detection limit estimator for multivariate calibration by an extension of the IUPAC recommendations for univariate methods. Analyst 2008, 133, 532–539. [Google Scholar] [CrossRef]
- Favaro, M.; Jeong, B.; Ross, P.N.; Yano, J.; Hussain, Z.; Liu, Z.; Crumlin, E.J. Unravelling the electrochemical double layer by direct probing of the solid/liquid interface. Nat. Commun. 2016, 7, 12695. [Google Scholar] [CrossRef]
- Feng, J.; Liu, K.; Graf, M.; Lihter, M.; Bulushev, R.D.; Dumcenco, D.; Alexander, D.T.; Krasnozhon, D.; Vuletic, T.; Kis, A.; et al. Electrochemical reaction in single layer MoS2: Nanopores opened atom by atom. Nano Lett. 2015, 15, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Nasir, M.Z.M.; Sofer, Z.; Ambrosi, A.; Pumera, M. A limited anodic and cathodic potential window of MoS2: Limitations in electrochemical applications. Nanoscale 2015, 7, 3126–3129. [Google Scholar] [CrossRef]
- Geng, X.; Sun, W.; Wu, W.; Chen, B.; Al-Hilo, A.; Benamara, M.; Zhu, H.; Watanabe, F.; Cui, J.; Chen, T.P. Pure and stable metallic phase molybdenum disulfide nanosheets for hydrogen evolution reaction. Nat. Commun. 2016, 7, 10672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.H.; Joo, M.-K.; Neumann, M.; Kim, H.; Lee, Y.H. Probing defect dynamics in monolayer MoS2 via noise nanospectroscopy. Nat. Commun. 2017, 8, 2121. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Fan, P.; Ke, X.; Xu, Y.; Huang, K.; Wei, H.; Yu, W.; Zhang, H.; Zhong, M.; Wu, H.; et al. Defective molybdenum sulfide quantum dots as highly active hydrogen evolution electrocatalysts. Nano Res. 2018, 11, 751–761. [Google Scholar] [CrossRef]
- KC, S.; Longo, R.C.; Wallace, R.M.; Cho, K. Surface oxidation energetics and kinetics on MoS2 monolayer. J. Appl. Phys. 2015, 117, 135301. [Google Scholar] [CrossRef]
- Baker, T.A.; Friend, C.M.; Kaxiras, E. Chlorine interaction with defects on the Au(111) surface: A first-principles theoretical investigation. J. Chem. Phys. 2008, 129, 104702. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Gold atoms stabilized on various supports catalyze the water-gas shift reaction. Acc. Chem. Res. 2014, 47, 783–792. [Google Scholar] [CrossRef]
- Kanbay, M.; Jensen, T.; Solak, Y.; Le, M.; Roncal-Jimenez, C.; Rivard, C.; Lanaspa, M.A.; Nakagawa, T.; Johnson, R.J. Uric acid in metabolic syndrome: From an innocent bystander to a central player. Eur. J. Intern. Med. 2016, 29, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Sulaiman Rahman, H. Antioxidant and oxidative stress: A mutual interplay in age-related diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Fischer, O.; Fischerová, E. Basic principles of voltammetry. In Experimental Techniques in Bioelectrochemistry; Brabec, V., Walz, D., Milazzo, G., Eds.; Birkhäuser: Basel, Switzerland, 1995; pp. 41–157. [Google Scholar]
- Govind Rajan, A.; Sresht, V.; Pádua, A.A.H.; Strano, M.S.; Blankschtein, D. Dominance of dispersion interactions and entropy over electrostatics in determining the wettability and friction of two-dimensional MoS2 surfaces. ACS Nano 2016, 10, 9145–9155. [Google Scholar] [CrossRef]
- Rännar, S.; Lindgren, F.; Geladi, P.; Wold, S. A PLS kernel algorithm for data sets with many variables and fewer objects. Part 1: Theory and algorithm. J. Chemom. 1994, 8, 111–125. [Google Scholar] [CrossRef]
- Tarpey, T. A note on the prediction sum of squares statistic for restricted least squares. Am. Stat. 2000, 54, 116–118. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazelles, R.; Shukla, R.P.; Ware, R.E.; Vinks, A.A.; Ben-Yoav, H. Electrochemical Determination of Hydroxyurea in a Complex Biological Matrix Using MoS2-Modified Electrodes and Chemometrics. Biomedicines 2021, 9, 6. https://doi.org/10.3390/biomedicines9010006
Cazelles R, Shukla RP, Ware RE, Vinks AA, Ben-Yoav H. Electrochemical Determination of Hydroxyurea in a Complex Biological Matrix Using MoS2-Modified Electrodes and Chemometrics. Biomedicines. 2021; 9(1):6. https://doi.org/10.3390/biomedicines9010006
Chicago/Turabian StyleCazelles, Remi, Rajendra P. Shukla, Russell E. Ware, Alexander A. Vinks, and Hadar Ben-Yoav. 2021. "Electrochemical Determination of Hydroxyurea in a Complex Biological Matrix Using MoS2-Modified Electrodes and Chemometrics" Biomedicines 9, no. 1: 6. https://doi.org/10.3390/biomedicines9010006