
Candida albicans Induces Foaming and Inflammation in Macrophages through FABP4: Its Implication for Atherosclerosis

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Macrophage Differentiation
2.3. Generation of Heat-Killed C. albicans
2.4. Cell Stimulation
2.5. Quantitative Real-Time PCR
2.6. Flow Cytometry: Staining of Cell-Surface Markers
2.7. Flow Cytometry: Intracellular Staining
2.8. Sandwich Enzyme-Linked Immunosorbent Assay
2.9. Small Interfering RNA Transfection
2.10. Nile Red Staining of Lipids
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. HKCA Stimulation Induced Foaming and Inflammation in Human Macrophages
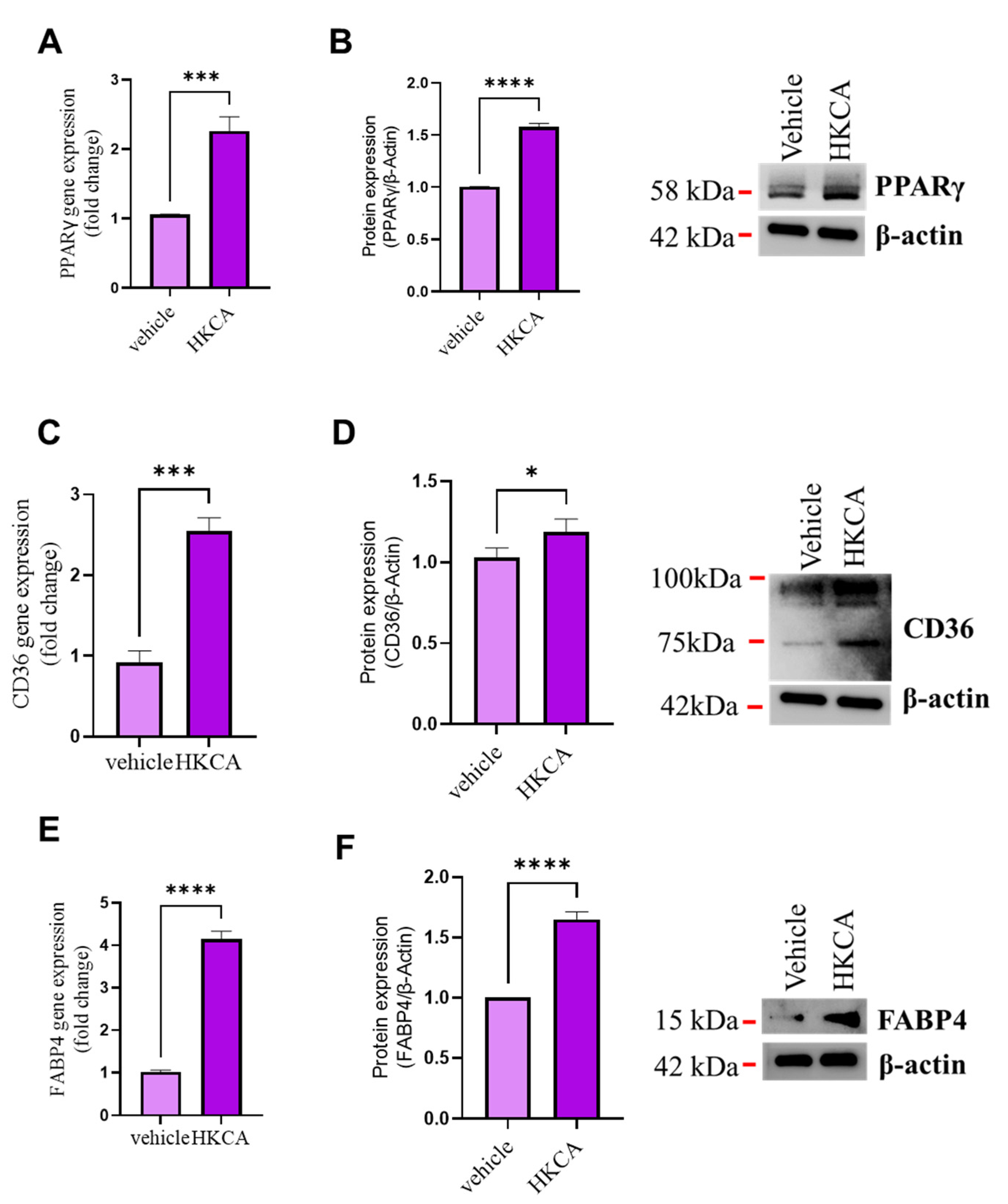
3.2. HKCA Stimulation Upregulates the Expression of CD36 and FABP4 in Foamy Macrophages
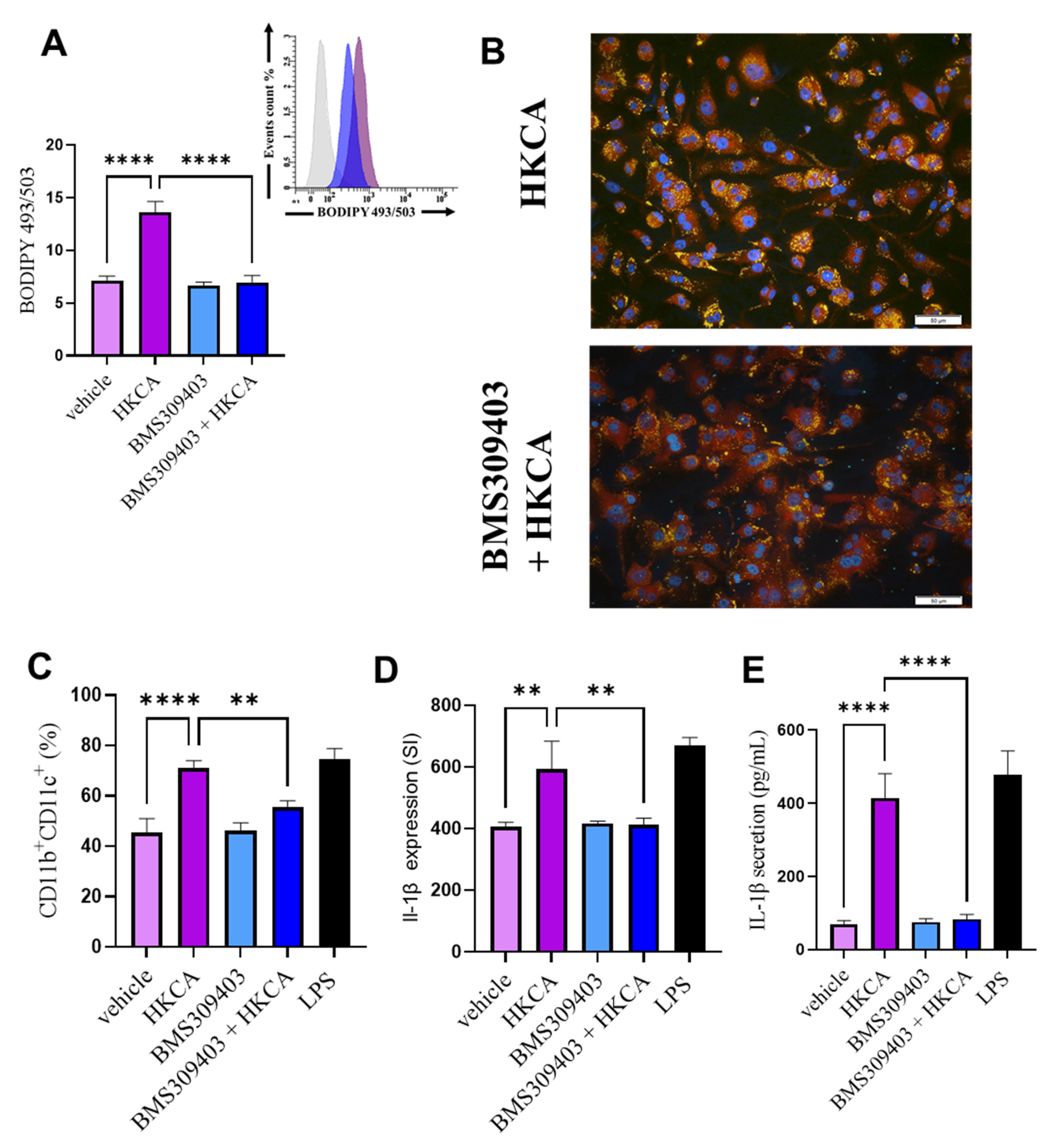
3.3. FABP4 Inhibition Prevents HKCA-Induced Macrophage Foaming and Associated Inflammation
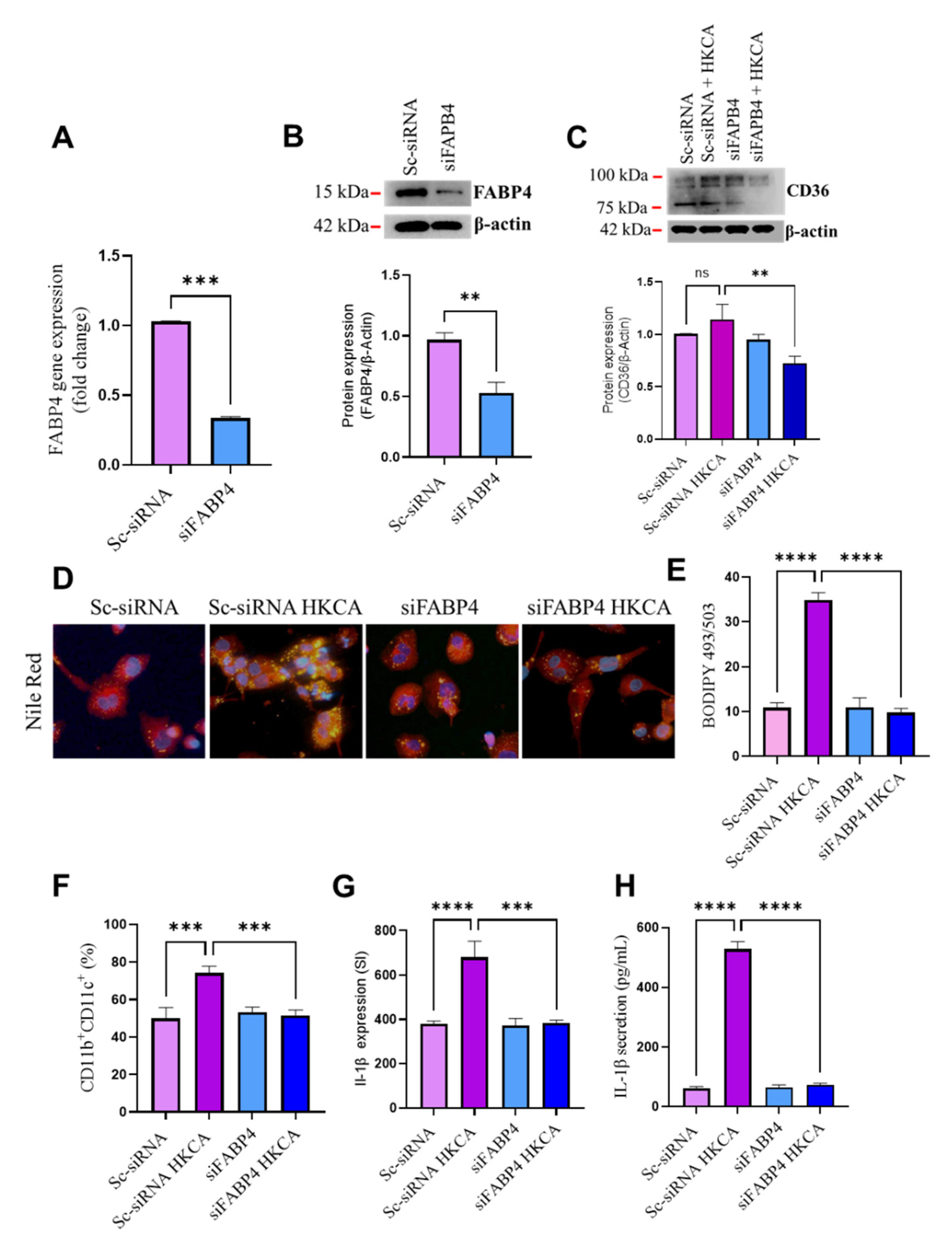
3.4. FABP4 Deficiency Suppresses HKCA-Induced Macrophage Foaming and Inflammation
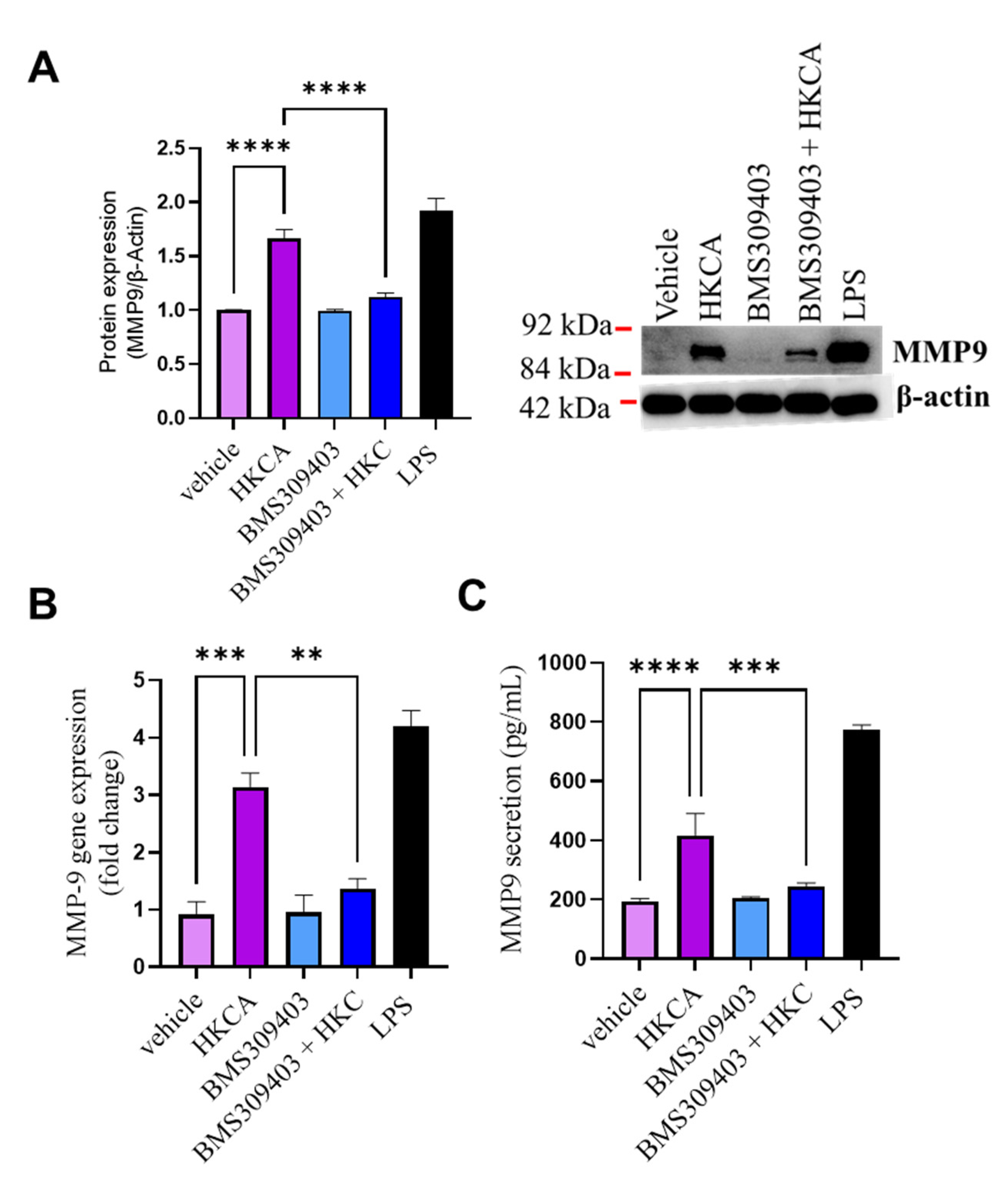
3.5. Effect of FABP4 Inhibition on HKCA-Induced Production of MMP-9
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagi-Miura, N.; Harada, T.; Shinohara, H.; Kurihara, K.; Adachi, Y.; Ishida-Okawara, A.; Oharaseki, T.; Takahashi, K.; Naoe, S.; Suzuki, K.; et al. Lethal and severe coronary arteritis in DBA/2 mice induced by fungal pathogen, CAWS, Candida albicans water-soluble fraction. Atherosclerosis 2006, 186, 310–320. [Google Scholar] [CrossRef]
- Ott, S.J.; El Mokhtari, N.E.; Rehman, A.; Rosenstiel, P.; Hellmig, S.; Kühbacher, T.; Lins, M.; Simon, R.; Schreiber, S. Fungal rDNA signatures in coronary atherosclerotic plaques. Environ. Microbiol. 2007, 9, 3035–3045. [Google Scholar] [CrossRef]
- Hall, C.J.; Dizcfalusy, U.; Sandstedt, K.; Bouhafs, L. Cryptococcus neoformans Causes Lipid Peroxidation; Therefore, It Is a Potential Inducer of Atherogenesis. Atherosclerosis 2011, 12, 92. [Google Scholar] [CrossRef]
- Epstein, S.E.; Zhou, Y.F.; Zhu, J. Infection and atherosclerosis: Emerging mechanistic paradigms. Circulation 1999, 100, e20–e28. [Google Scholar] [CrossRef]
- Mangge, H.; Almer, G. Immune-Mediated Inflammation in Vulnerable Atherosclerotic Plaques. Molecules 2019, 24, 3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikane, Y.; Koga, M.; Imanaka-Yoshida, K.; Cho, T.; Yamamoto, Y.; Yoshida, T.; Hashimoto, J.; Hirose, S.; Yoshimura, K. JNK is critical for the development of Candida albicans-induced vascular lesions in a mouse model of Kawasaki Disease. Cardiovasc. Pathol. 2015, 24, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, O.; Shahzadi, M.; Kordbacheh, P.; Zaini, F.; Mahmoudi, S.; Mahmoudi, M.; Bahreini, H.; Safara, M.; Mirhendi, H. Detection of Fungal Elements in Atherosclerotic Plaques Using Mycological, Pathological and Molecular Methods. Iran. J. Public Health 2015, 44, 1121–1125. [Google Scholar]
- Nurgeldiyeva, M.J.; Hojakuliyev, B.G.; Muhammedov, M.B. Correlation of atherogenesis with an infection of Candida albicans. Int. J. Clin. Exp. Med. 2014, 7, 2137–2143. [Google Scholar]
- Yeter, H.H.; Erten, Y.; Sevmez, H.; Korucu, B.; Kalkanci, A.; Elbeg, S.; Altok, K.; Bali, M.; Yilmaz, H. Oral Candida Colonization as a Risk Factor for Chronic Inflammation and Atherosclerosis in Hemodialysis Patients. Ther. Apher. Dial. 2019, 23, 542–549. [Google Scholar] [CrossRef]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Grajchen, E.; Hendriks, J.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef]
- Russell, D.G.; Cardona, P.-J.; Kim, M.-J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rashed, F.; Ahmad, Z.; Iskandar, M.A.; Tuomilehto, J.; Al-Mulla, F.; Ahmad, R. TNF-α Induces a Pro-Inflammatory Phenotypic Shift in Monocytes through ACSL1: Relevance to Metabolic Inflammation. Cell. Physiol. Biochem. 2019, 52, 397–407. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral sphingomyelinase 2 regulates inflammatory responses in monocytes/macrophages induced by TNF-α. Sci. Rep. 2020, 10, 16802. [Google Scholar] [CrossRef]
- Gatto, F.; Cagliani, R.; Catelani, T.; Guarnieri, D.; Moglianetti, M.; Pompa, P.P.; Bardi, G. PMA-Induced THP-1 Macrophage Differentiation is Not Impaired by Citrate-Coated Platinum Nanoparticles. Nanomaterials 2017, 7, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rashed, F.; Ahmad, Z.; Snider, A.J.; Thomas, R.; Kochumon, S.; Melhem, M.; Sindhu, S.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; et al. Ceramide kinase regulates TNF-α-induced immune responses in human monocytic cells. Sci. Rep. 2021, 11, 8259. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Thomas, R.; Al-Roub, A.; Al-Mulla, F.; Ahmad, R. LPS Induces GM-CSF Production by Breast Cancer MDA-MB-231 Cells via Long-Chain Acyl-CoA Synthetase 1. Molecules 2020, 25, 4709. [Google Scholar] [CrossRef] [PubMed]
- Wray, G.M.; Foster, S.J.; Hinds, C.J.; Thiemermann, C. A cell wall component from pathogenic and non-pathogenic gram-positive bacteria (peptidoglycan) synergises with endotoxin to cause the release of tumour necrosis factor-α, nitric oxide production, shock, and multiple organ injury/dysfunction in the rat. Shock 2001, 15, 135–142. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Kochumon, S.; Usmani, S.; Sindhu, S.; Ahmad, R. Pam3CSK4 Induces MMP-9 Expression in Human Monocytic THP-1 Cells. Cell. Physiol. Biochem. 2017, 41, 1993–2003. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Rashed, F.; Akhter, N.; Al-Mulla, F.; Ahmad, R. ACSL1 Regulates TNFalpha-Induced GM-CSF Production by Breast Cancer MDA-MB-231 Cells. Biomolecules 2019, 9, 555. [Google Scholar] [CrossRef] [Green Version]
- Al-Rashed, F.; Sindhu, S.; Al Madhoun, A.; Ahmad, Z.; AlMekhled, D.; Azim, R.; Al-Kandari, S.; Wahid, M.A.-A.; Al-Mulla, F.; Ahmad, R. Elevated resting heart rate as a predictor of inflammation and cardiovascular risk in healthy obese individuals. Sci. Rep. 2021, 11, 13883. [Google Scholar] [CrossRef]
- Kochumon, S.; Arefanian, H.; Azim, R.; Shenouda, S.; Jacob, T.; Abu Khalaf, N.; Al-Rashed, F.; Hasan, A.; Sindhu, S.; Al-Mulla, F.; et al. Stearic Acid and TNF-α Co-Operatively Potentiate MIP-1α Production in Monocytic Cells via MyD88 Independent TLR4/TBK/IRF3 Signaling Pathway. Biomedicines 2020, 8, 403. [Google Scholar] [CrossRef]
- Al-Roub, A.; Akhter, N.; Al-Sayyar, A.; Wilson, A.; Thomas, R.; Kochumon, S.; Al-Rashed, F.; Al-Mulla, F.; Sindhu, S.; Ahmad, R. Short Chain Fatty Acid Acetate Increases TNFα-Induced MCP-1 Production in Monocytic Cells via ACSL1/MAPK/NF-κB Axis. Int. J. Mol. Sci. 2021, 22, 7683. [Google Scholar] [CrossRef]
- Cole, T.A.; Fok, A.K.; Ueno, M.S.; Allen, R.D. Use of nile red as a rapid measure of lipid content in ciliates. Eur. J. Protistol. 1990, 25, 361–368. [Google Scholar] [CrossRef]
- Biswas, S.; Gao, D.; Altemus, J.B.; Rekhi, U.R.; Chang, E.; Febbraio, M.; Byzova, T.V.; Podrez, E.A. Circulating CD36 is increased in hyperlipidemic mice: Cellular sources and triggers of release. Free Radic. Biol. Med. 2021, 168, 180–188. [Google Scholar] [CrossRef]
- Boß, M.; Kemmerer, M.; Brüne, B.; Namgaladze, D. FABP4 inhibition suppresses PPARγ activity and VLDL-induced foam cell formation in IL-4-polarized human macrophages. Atherosclerosis 2015, 240, 424–430. [Google Scholar] [CrossRef]
- Xu, H.; Hertzel, A.V.; Steen, K.A.; Wang, Q.; Suttles, J.; Bernlohr, D.A. Uncoupling Lipid Metabolism from Inflammation through Fatty Acid Binding Protein-Dependent Expression of UCP2. Mol. Cell. Biol. 2015, 35, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Liu, L.; Yin, L.; Xu, L.; Tang, Z.; Qi, Y.; Mao, Z.; Zhao, Y.; Ma, X.; Peng, J. Retracted Article: FABP4 contributes to renal interstitial fibrosis via mediating inflammation and lipid metabolism. Cell Death Dis. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Lan, H.; Cheng, C.C.; Kowalski, T.J.; Pang, L.; Shan, L.; Chuang, C.C.; Jackson, J.; Rojas-Triana, A.; Bober, L.; Liu, L.; et al. Small-molecule inhibitors of FABP4/5 ameliorate dyslipidemia but not insulin resistance in mice with diet-induced obesity. J. Lipid Res. 2011, 52, 646–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caimi, G.; Hopps, E.; Montana, M.; Urso, C.; Carollo, C.; Canino, B.; Presti, R.L. The function of matrix metalloproteinase-9 (MMP-9) and its tissue inhibitor (TIMP-1) in several clinical conditions: Results and analysis of our survey. Clin. Hemorheol. Microcirc. 2021, 78, 401–416. [Google Scholar] [CrossRef]
- Głogowska-Ligus, J.; Dąbek, J.; Piechota, M.; Gallert-Kopyto, W.; Lepich, T.; Korzeń, D.; Gąsior, Z. Can the expression of the metalloproteinase 9 gene and its inhibitor be considered as markers of heart failure? Minerva Cardiol. Angiol. 2020, 69, 172–177. [Google Scholar] [PubMed]
- Wågsäter, D.; Zhu, C.; Björkegren, J.; Skogsberg, J.; Eriksson, P. MMP-2 and MMP-9 are prominent matrix metalloproteinases during atherosclerosis development in the Ldlr(-/-)Apob(100/100) mouse. Int. J. Mol. Med. 2011, 28, 247–253. [Google Scholar]
- Recuero-Checa, M.A.; Sharma, M.; Lau, C.; Watkins, P.A.; Gaydos, C.A.; Dean, D. Chlamydia trachomatis growth and development requires the activity of host Long-chain Acyl-CoA Synthetases (ACSLs). Sci. Rep. 2016, 6, 23148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Rho, J.; Woo, B.; Joo, J.; Song, J.; Park, H.; Lee, J.; Lee, J. Periodontal Pathogens Modulate Lipid Flux via Fatty Acid Binding Protein 4. J. Dent. Res. 2019, 98, 1511–1520. [Google Scholar] [CrossRef]
- Gómez-Díaz, R.A.; Ramírez-Soriano, E.; Hajj, J.T.; Cruz, E.B.; Galicia, C.J.; Villasís-Keever, M.Á.; Aguilar-Salinas, C.A.; Wacher, N.H. Association between carotid intima-media thickness, buccodental status, and glycemic control in pediatric type 1 diabetes. Pediatr. Diabetes 2012, 13, 552–558. [Google Scholar] [CrossRef]
- Wu, H.; Gower, R.M.; Wang, H.; Perrard, X.-Y.D.; Ma, R.; Bullard, D.C.; Burns, A.R.; Paul, A.; Smith, C.W.; Simon, S.I.; et al. Functional Role of CD11c + Monocytes in Atherogenesis Associated with Hypercholesterolemia. Circulation 2009, 119, 2708–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Zaitsev, K.; Kim, K.-W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G.; et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Dkhar, H.K.; Chandra, V.; Dave, S.; Nanduri, R.; Janmeja, A.K.; Agrewala, J.N.; Gupta, P. Mycobacterium tuberculosis Modulates Macrophage Lipid-Sensing Nuclear Receptors PPARγ and TR4 for Survival. J. Immunol. 2012, 188, 5593–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, P.E.; Roque, N.R.; Magalhaes, K.G.; Mattos, K.A.; Teixeira, L.; Maya-Monteiro, C.; Almeida, C.J.; Castro-Faria-Neto, H.C.; Ryffel, B.; Quesniaux, V.F.; et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1841, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.A.A.; et al. Peroxisome proliferator-activated receptor-γ in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas Bervejillo, M.; Bonanata, J.; Franchini, G.R.; Richeri, A.; Marqués, J.M.; Freeman, B.A.; Schopfer, F.J.; Coitiño, E.L.; Córsico, B.; Rubbo, H.; et al. A FABP4-PPARγ signaling axis regulates human monocyte responses to electrophilic fatty acid nitroalkenes. Redox Biol. 2020, 29, 101376. [Google Scholar] [CrossRef]
- Tan, N.S.; Shaw, N.S.; Vinckenbosch, N.; Liu, P.; Yasmin, R.; Desvergne, B.; Wahli, W.; Noy, N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 2002, 22, 5114–5127. [Google Scholar] [CrossRef] [Green Version]
- Garin-Shkolnik, T.; Rudich, A.; Hotamisligil, G.S.; Rubinstein, M. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. Diabetes 2014, 63, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Chistiakov, D.A.; Grechko, A.; Orekhov, A.N. Matrix metalloproteinases in pro-atherosclerotic arterial remodeling. J. Mol. Cell. Cardiol. 2018, 123, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Stabouli, S.; Kotsis, V.; Maliachova, O.; Printza, N.; Chainoglou, A.; Christoforidis, A.; Taparkou, A.; Dotis, J.; Farmaki, E.; Zafeiriou, D. Matrix metalloproteinase −2, −9 and arterial stiffness in children and adolescents: The role of chronic kidney disease, diabetes, and hypertension. Int. J. Cardiol. Hypertens. 2020, 4, 100025. [Google Scholar] [CrossRef]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Pärnänen, P.; Meurman, J.; Sorsa, T. The effects of Candida proteinases on human proMMP-9, TIMP-1 and TIMP-2. Mycoses 2010, 54, 325–330. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, M.; Al-Rashed, F.; Albaqsumi, Z.; Alobaid, K.; Alqabandi, R.; Al-Mulla, F.; Ahmad, R. Candida albicans Induces Foaming and Inflammation in Macrophages through FABP4: Its Implication for Atherosclerosis. Biomedicines 2021, 9, 1567. https://doi.org/10.3390/biomedicines9111567
Haider M, Al-Rashed F, Albaqsumi Z, Alobaid K, Alqabandi R, Al-Mulla F, Ahmad R. Candida albicans Induces Foaming and Inflammation in Macrophages through FABP4: Its Implication for Atherosclerosis. Biomedicines. 2021; 9(11):1567. https://doi.org/10.3390/biomedicines9111567
Chicago/Turabian StyleHaider, Mohammed, Fatema Al-Rashed, Zahraa Albaqsumi, Khaled Alobaid, Rawan Alqabandi, Fahd Al-Mulla, and Rasheed Ahmad. 2021. "Candida albicans Induces Foaming and Inflammation in Macrophages through FABP4: Its Implication for Atherosclerosis" Biomedicines 9, no. 11: 1567. https://doi.org/10.3390/biomedicines9111567
APA StyleHaider, M., Al-Rashed, F., Albaqsumi, Z., Alobaid, K., Alqabandi, R., Al-Mulla, F., & Ahmad, R. (2021). Candida albicans Induces Foaming and Inflammation in Macrophages through FABP4: Its Implication for Atherosclerosis. Biomedicines, 9(11), 1567. https://doi.org/10.3390/biomedicines9111567