
The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target


Abstract
1. Introduction
2. Functional Dynamics of the HBV Core Protein and Capsid in Virus Replication
2.1. Cp in Early Steps of the HBV Infection Cycle
2.2. Cp and HBV cccDNA
2.3. De novo Viral Protein Synthesis and Cp Functions in Progeny Virion Production
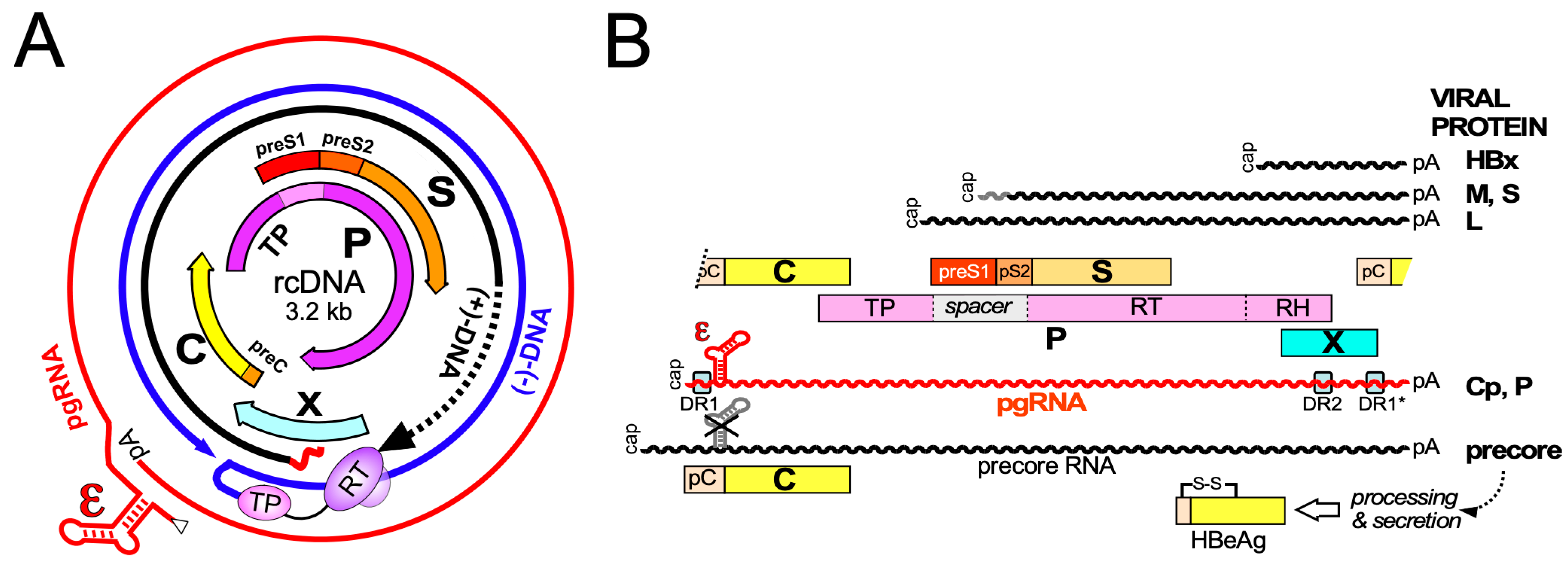
3. Primary Structure and Biochemical Dynamics of HBV Core Proteins
3.1. Domain Structure of HBV Cp
3.2. HBV Precore Protein—A Nonassembling Secretory Cp Derivative
3.3. Posttranslational Modifications of Cp
3.4. Dynamic Cp Phosphorylation/Dephosphorylation for Regulated Nucleic Acid Binding
4. Overall Structural Dynamics of HBV Cp
4.1. Early Evidence for Autonomous Cp Self-Assembly into Capsid-Like Particles
T = 3 vs. T = 4 Dimorphism of HBV Cp Capsids
4.2. High-Resolution Structure Determination of HBV Capsids
Crystal-Independent High-Resolution Cp Analysis
4.3. Monitoring Assembly Dynamics
4.3.1. Disassembly–Reassembly Studies
4.3.2. Specialized Techniques to Study Cp Assembly
4.3.3. Cp Assembly Intermediates
4.3.4. Specific Versus Non-Specific RNA as a Cofactor in HBV Nucleocapsid Assembly
5. Targeting HBV Capsid Dynamics
5.1. Viral Structural Proteins as Therapeutic Targets
5.2. Discovery of the First and Newer HBV Cp Targeting Compounds
5.3. Towards Understanding the Mechanisms of CAM Action
5.3.1. Biophysical/Structural Characterization of HBV Cp-CAM Interactions
5.3.2. Preclinical Assessment of CAM Anti-HBV Activity
5.3.3. CAMs in Clinical Trials
5.3.4. CAMs as Combination Therapy Components
5.4. CAM Resistance—A Relevant Therapeutic Issue to Be Addressed
6. Conclusions, Open Questions, and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Hepatitis B Fact Sheet. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 21 September 2021).
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608. [Google Scholar] [CrossRef]
- Buti, M.; Marcos-Fosch, C.; Esteban, R. Nucleos(t)ide analogue therapy: The role of tenofovir alafenamide. Liver Int. 2021, 41 (Suppl. 1), 9–14. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Jeng, W.J.; Lok, A.S.F. Is cure of hepatitis B infection a mission possible? In Hepatitis B Virus and Liver Disease; Kao, J.-H., Ed.; Springer: Singapore, 2021; pp. 475–495. [Google Scholar]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Gao, L.; Han, X.; Hu, T.; Hu, Y.; Liu, H.; Thomas, A.W.; Yan, Z.; Yang, S.; Young, J.A.T.; et al. Discovery of Small Molecule Therapeutics for Treatment of Chronic HBV Infection. ACS Infect. Dis. 2018, 4, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Prifti, G.M.; Moianos, D.; Giannakopoulou, E.; Pardali, V.; Tavis, J.E.; Zoidis, G. Recent Advances in Hepatitis B Treatment. Pharm. 2021, 14, 417. [Google Scholar] [CrossRef]
- Ligat, G.; Verrier, E.R.; Nassal, M.; Baumert, T.F. Hepatitis B virus-host interactions and novel targets for viral cure. Curr. Opin. Virol. 2021, 49, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Le Bert, N. Immunotherapy for Chronic Hepatitis B Virus Infection. Gut Liver 2018, 12, 497–507. [Google Scholar] [CrossRef]
- Lang, J.; Neumann-Haefelin, C.; Thimme, R. Immunological cure of HBV infection. Hepatol. Int. 2019, 13, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.J.; Protzer, U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology 2019, 156, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, P.; Schlicksup, C.J.; Zlotnick, A.; Jacobson, S.C. Analytical Techniques to Characterize the Structure, Properties, and Assembly of Virus Capsids. Anal. Chem. 2019, 91, 622–636. [Google Scholar] [CrossRef]
- Viswanathan, U.; Mani, N.; Hu, Z.; Ban, H.; Du, Y.; Hu, J.; Chang, J.; Guo, J.T. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antivir. Res. 2020, 182, 104917. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 2016, 64, S32–S40. [Google Scholar] [CrossRef]
- Tu, T.; Urban, S. Virus entry and its inhibition to prevent and treat hepatitis B and hepatitis D virus infections. Curr. Opin. Virol. 2018, 30, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis D virus in 2021: Virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut 2021. [Google Scholar] [CrossRef]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef]
- Osseman, Q.; Gallucci, L.; Au, S.; Cazenave, C.; Berdance, E.; Blondot, M.L.; Cassany, A.; Begu, D.; Ragues, J.; Aknin, C.; et al. The chaperone dynein LL1 mediates cytoplasmic transport of empty and mature hepatitis B virus capsids. J. Hepatol. 2018, 68, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, L.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Nassal, M. A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation-and beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef]
- Marchetti, A.L.; Guo, H. New Insights on Molecular Mechanism of Hepatitis B Virus Covalently Closed Circular DNA Formation. Cells 2020, 9, 2430. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog 2010, 6, e1001162. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liang, C.; Li, F.; Wang, L.; Wu, X.; Lu, A.; Xiao, G.; Zhang, G. The Rules and Functions of Nucleocytoplasmic Shuttling Proteins. Int. J. Mol. Sci. 2018, 19, 1445. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Zlotnick, A. HBV Core Protein Is in Flux between Cytoplasmic, Nuclear, and Nucleolar Compartments. mBio 2021, 12. [Google Scholar] [CrossRef]
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021, 75, 706–717. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and Characterizing Interplay between Hepatitis B Virus X Protein and Smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef]
- Shen, C.; Feng, X.; Mao, T.; Yang, D.; Zou, J.; Zao, X.; Deng, Q.; Chen, X.; Lu, F. Yin-Yang 1 and HBx protein activate HBV transcription by mediating the spatial interaction of cccDNA minichromosome with cellular chromosome 19p13.11. Emerg. Microbes Infect. 2020, 9, 2455–2464. [Google Scholar] [CrossRef]
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell Rep. 2021, 35, 109288. [Google Scholar] [CrossRef]
- Allweiss, L.; Giersch, K.; Pirosu, A.; Volz, T.; Muench, R.C.; Beran, R.K.; Urban, S.; Javanbakht, H.; Fletcher, S.P.; Lutgehetmann, M.; et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.T.; Hu, J.L.; Ren, J.H.; Yu, H.B.; Zhong, S.; Wai Wong, V.K.; Kwan Law, B.Y.; Chen, W.X.; Xu, H.M.; Zhang, Z.Z.; et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx. J. Hepatol. 2021, 74, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Chabrolles, H.; Auclair, H.; Vegna, S.; Lahlali, T.; Pons, C.; Michelet, M.; Coute, Y.; Belmudes, L.; Chadeuf, G.; Kim, Y.; et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog. 2020, 16, e1008593. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Pastor, F.; Charles, E.; Pons, C.; Auclair, H.; Fusil, F.; Rivoire, M.; Cosset, F.L.; Durantel, D.; Salvetti, A. Evidence for long-term association of virion-delivered hepatitis B virus core protein with cccDNA independently of viral protein production. J. Hepatol. Rep. 2021. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase kappa Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2021, 3, 100195. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Makbul, C.; Nassal, M.; Böttcher, B. Slowly folding surface extension in the prototypic avian hepatitis B virus capsid governs stability. eLife 2020, 9. [Google Scholar] [CrossRef]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- DiMattia, M.A.; Watts, N.R.; Stahl, S.J.; Grimes, J.M.; Steven, A.C.; Stuart, D.I.; Wingfield, P.T. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure 2013, 21, 133–142. [Google Scholar] [CrossRef]
- Nassal, M.; Rieger, A. An intramolecular disulfide bridge between Cys-7 and Cys61 determines the structure of the secretory core gene product (e antigen) of hepatitis B virus. J. Virol. 1993, 67, 4307–4315. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M.; Junker-Niepmann, M.; Schaller, H. Translational inactivation of RNA function: Discrimination against a subset of genomic transcripts during HBV nucleocapsid assembly. Cell 1990, 63, 1357–1363. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Seitz, S.; Lauber, C.; Nassal, M. Conservation of the HBV RNA element epsilon in nackednaviruses reveals ancient origin of protein-primed reverse transcription. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef]
- Meier, M.A.; Calabrese, D.; Suslov, A.; Terracciano, L.M.; Heim, M.H.; Wieland, S. Ubiquitous expression of HBsAg from integrated HBV DNA in patients with low viral load. J. Hepatol. 2021, 75, 840–847. [Google Scholar] [CrossRef]
- Ho, J.K.; Jeevan-Raj, B.; Netter, H.J. Hepatitis B Virus (HBV) Subviral Particles as Protective Vaccines and Vaccine Platforms. Viruses 2020, 12, 126. [Google Scholar] [CrossRef]
- Vaillant, A. HBsAg, Subviral Particles, and Their Clearance in Establishing a Functional Cure of Chronic Hepatitis B Virus Infection. ACS Infect. Dis. 2021, 7, 1351–1368. [Google Scholar] [CrossRef]
- Zeyen, L.; Doring, T.; Stieler, J.T.; Prange, R. Hepatitis B subviral envelope particles use the COPII machinery for intracellular transport via selective exploitation of Sec24A and Sec23B. Cell Microbiol. 2020, 22, e13181. [Google Scholar] [CrossRef]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2015, 90, 3330–3341. [Google Scholar] [CrossRef]
- Zeyen, L.; Prange, R. Host Cell Rab GTPases in Hepatitis B Virus Infection. Front. Cell Dev. Biol. 2018, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Habjanic, J.; Schutz, A.K.; Bartenschlager, R. The Hepatitis B Virus Envelope Proteins: Molecular Gymnastics Throughout the Viral Life Cycle. Annu. Rev. Virol. 2020, 7, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Iancu, C.; Volz, T.; Mier, W.; Dandri, M.; Urban, S.; Bartenschlager, R. A Slow Maturation Process Renders Hepatitis B Virus Infectious. Cell Host Microbe 2016, 20, 25–35. [Google Scholar] [CrossRef]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Böttcher, B.; Nassal, M. Structure of Mutant Hepatitis B Core Protein Capsids with Premature Secretion Phenotype. J. Mol. Biol. 2018, 430, 4941–4954. [Google Scholar] [CrossRef]
- Yuan, T.T.; Sahu, G.K.; Whitehead, W.E.; Greenberg, R.; Shih, C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 1999, 73, 5731–5740. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef]
- Ning, X.; Luckenbaugh, L.; Liu, K.; Bruss, V.; Sureau, C.; Hu, J. Common and Distinct Capsid and Surface Protein Requirements for Secretion of Complete and Genome-Free Hepatitis B Virions. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Lecoq, L.; Wang, S.; Dujardin, M.; Zimmermann, P.; Schuster, L.; Fogeron, M.L.; Briday, M.; Schledorn, M.; Wiegand, T.; Cole, L.; et al. A pocket-factor-triggered conformational switch in the hepatitis B virus capsid. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Bardens, A.; Doring, T.; Stieler, J.; Prange, R. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT-independent manner. Cell Microbiol. 2011, 13, 602–619. [Google Scholar] [CrossRef]
- Chou, S.F.; Tsai, M.L.; Huang, J.Y.; Chang, Y.S.; Shih, C. The Dual Role of an ESCRT-0 Component HGS in HBV Transcription and Naked Capsid Secretion. PLoS Pathog. 2015, 11, e1005123. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, B.; Valdes, J.D.; Sun, J.; Guo, H. Serum Hepatitis B Virus RNA: A New Potential Biomarker for Chronic Hepatitis B Virus Infection. Hepatology 2019, 69, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xie, Z.; Cai, D.; Yu, X.; Zhang, H.; Kim, E.S.; Zhou, B.; Hou, J.; Zhang, X.; Huang, Q.; et al. Biogenesis and molecular characteristics of serum hepatitis B virus RNA. PLoS Pathog. 2020, 16, e1008945. [Google Scholar] [CrossRef]
- Den Boon, J.A.; Ahlquist, P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Mann, C.C.; Hornung, V. Molecular mechanisms of nonself nucleic acid recognition by the innate immune system. Eur. J. Immunol. 2021, 51, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, N.; Li, Z.; Xu, G.; Zhan, X.; Tang, J.; Xiao, X.; Bai, Z. The Cytosolic DNA-Sensing cGAS-STING Pathway in Liver Diseases. Front. Cell Dev. Biol. 2021, 9, 717610. [Google Scholar] [CrossRef] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e1722. [Google Scholar] [CrossRef]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere with Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef]
- Birnbaum, F.; Nassal, M. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [Google Scholar] [CrossRef]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef]
- Liu, H.; Xi, J.; Hu, J. Regulation of Hepatitis B Virus Replication by Cyclin Docking Motifs in Core Protein. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Liu, K.; Luckenbaugh, L.; Ning, X.; Xi, J.; Hu, J. Multiple roles of core protein linker in hepatitis B virus replication. PLoS Pathog. 2018, 14, e1007085. [Google Scholar] [CrossRef]
- Xi, J.; Luckenbaugh, L.; Hu, J. Multiple roles of PP2A binding motif in hepatitis B virus core linker and PP2A in regulating core phosphorylation state and viral replication. PLoS Pathog. 2021, 17, e1009230. [Google Scholar] [CrossRef]
- Heger-Stevic, J.; Kolb, P.; Walker, A.; Nassal, M. Displaying Whole-Chain Proteins on Hepatitis B Virus Capsid-Like Particles. Methods Mol. Biol. 2018, 1776, 503–531. [Google Scholar] [CrossRef]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Böttcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef]
- Lecoq, L.; Wang, S.; Wiegand, T.; Bressanelli, S.; Nassal, M.; Meier, B.H.; Bockmann, A. Solid-state [(13)C-(15)N] NMR resonance assignment of hepatitis B virus core protein. Biomol. NMR Assign. 2018, 12, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, L.; Wang, S.; Wiegand, T.; Bressanelli, S.; Nassal, M.; Meier, B.H.; Bockmann, A. Localizing Conformational Hinges by NMR: Where Do Hepatitis B Virus Core Proteins Adapt for Capsid Assembly? ChemPhysChem 2018, 19, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, W.H.; Glebe, D.; Kramvis, A.; Magnius, L.O. Peculiarities in the designations of hepatitis B virus genes, their products, and their antigenic specificities: A potential source of misunderstandings. Virus Genes 2020, 56, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qin, Y.; Zhang, J.; Jia, L.; Fu, S.; Wang, Y.; Li, J.; Tong, S. Tracing the evolutionary history of hepadnaviruses in terms of e antigen and middle envelope protein expression or processing. Virus Res. 2020, 276, 197825. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Luckenbaugh, L.; Perlman, D.; Revill, P.A.; Wieland, S.F.; Menne, S.; Hu, J. Characterization and Application of Precore/Core-Related Antigens in Animal Models of Hepatitis B Virus Infection. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Kanda, T.; Imazeki, F.; Nakamoto, S.; Tanaka, T.; Arai, M.; Roger, T.; Shirasawa, H.; Nomura, F.; Yokosuka, O. Hepatitis B virus e antigen physically associates with receptor-interacting serine/threonine protein kinase 2 and regulates IL-6 gene expression. J. Infect. Dis. 2012, 206, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Milich, D.R. Is the function of the HBeAg really unknown? Hum. Vaccin. Immunother. 2019, 15, 2187–2191. [Google Scholar] [CrossRef]
- Wasenauer, G.; Kock, J.; Schlicht, H.J. Relevance of cysteine residues for biosynthesis and antigenicity of human hepatitis B virus e protein. J. Virol. 1993, 67, 1315–1321. [Google Scholar] [CrossRef]
- Langerova, H.; Lubyova, B.; Zabransky, A.; Hubalek, M.; Glendova, K.; Aillot, L.; Hodek, J.; Strunin, D.; Janovec, V.; Hirsch, I.; et al. Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination. Cells 2020, 9, 2547. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Albin, C.; Robinson, W.S. Protein kinase activity in hepatitis B virus. J. Virol. 1980, 34, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, W.H.; Goldmann, U.; Muller, R.; Stibbe, W.; Wolff, W. Specificity and localization of the hepatitis B virus-associated protein kinase. J. Virol. 1982, 42, 761–766. [Google Scholar] [CrossRef]
- Pugh, J.; Zweidler, A.; Summers, J. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 1989, 63, 1371–1376. [Google Scholar] [CrossRef]
- Perlman, D.H.; Berg, E.A.; O’Connor P., B.; Costello, C.E.; Hu, J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. USA 2005, 102, 9020–9025. [Google Scholar] [CrossRef] [PubMed]
- Basagoudanavar, S.H.; Perlman, D.H.; Hu, J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 2007, 81, 1641–1649. [Google Scholar] [CrossRef]
- Lewellyn, E.B.; Loeb, D.D. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS ONE 2011, 6, e17202. [Google Scholar] [CrossRef]
- Chua, P.K.; Tang, F.M.; Huang, J.Y.; Suen, C.S.; Shih, C. Testing the balanced electrostatic interaction hypothesis of hepatitis B virus DNA synthesis by using an in vivo charge rebalance approach. J. Virol. 2010, 84, 2340–2351. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, P.Y.; Yang, C.J.; Chu, T.H.; Chang, C.H.; Chiang, C.; Tang, F.M.; Lee, C.Y.; Shih, C. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci. Rep. 2016, 6, 38959. [Google Scholar] [CrossRef]
- Porterfield, J.Z.; Dhason, M.S.; Loeb, D.D.; Nassal, M.; Stray, S.J.; Zlotnick, A. Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J. Virol. 2010, 84, 7174–7184. [Google Scholar] [CrossRef]
- Strods, A.; Ose, V.; Bogans, J.; Cielens, I.; Kalnins, G.; Radovica, I.; Kazaks, A.; Pumpens, P.; Renhofa, R. Preparation by alkaline treatment and detailed characterisation of empty hepatitis B virus core particles for vaccine and gene therapy applications. Sci. Rep. 2015, 5, 11639. [Google Scholar] [CrossRef]
- Porterfield, J.Z.; Zlotnick, A. A simple and general method for determining the protein and nucleic acid content of viruses by UV absorbance. Virology 2010, 407, 281–288. [Google Scholar] [CrossRef]
- Köck, J.; Rösler, C.; Zhang, J.J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Köck, J.; Nassal, M.; Deres, K.; Blum, H.E.; von Weizsäcker, F. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 2004, 78, 13812–13818. [Google Scholar] [CrossRef]
- Le Pogam, S.; Chua, P.K.; Newman, M.; Shih, C. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165-173). J. Virol. 2005, 79, 1871–1887. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Daub, H.; Blencke, S.; Habenberger, P.; Kurtenbach, A.; Dennenmoser, J.; Wissing, J.; Ullrich, A.; Cotten, M. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 2002, 76, 8124–8137. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xi, J.; Gao, L.; Hu, J. Role of Hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PLoS Pathog. 2020, 16, e1008459. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Basagoudanavar, S.H.; Liu, K.; Luckenbaugh, L.; Wei, D.; Wang, C.; Wei, B.; Zhao, Y.; Yan, T.; Delaney, W.; et al. Capsid Phosphorylation State and Hepadnavirus Virion Secretion. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, Z.; Cheng, J.; Wu, S.; Luo, Y.; Chang, J.; Hu, J.; Guo, J.T. Hepatitis B Virus Core Protein Dephosphorylation Occurs during Pregenomic RNA Encapsidation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ban, H.; Zheng, H.; Liu, M.; Chang, J.; Guo, J.T. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PLoS Pathog. 2020, 16, e1008669. [Google Scholar] [CrossRef]
- Nassal, M.; Rieger, A.; Steinau, O. Topological analysis of the hepatitis B virus core particle by cysteine-cysteine cross-linking. J. Mol. Biol. 1992, 225, 1013–1025. [Google Scholar] [CrossRef]
- Zhou, S.; Standring, D.N. Hepatitis B virus capsid particles are assembled from core-protein dimer precursors. Proc. Natl. Acad. Sci. USA 1992, 89, 10046–10050. [Google Scholar] [CrossRef]
- Seifer, M.; Zhou, S.; Standring, D.N. A micromolar pool of antigenically distinct precursors is required to initiate cooperative assembly of hepatitis B virus capsids in Xenopus oocytes. J. Virol. 1993, 67, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Kiselev, N.A.; Bottcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Crick, F.H.; Watson, J.D. Structure of small viruses. Nature 1956, 177, 473–475. [Google Scholar] [CrossRef]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef]
- Johnson, J.E.; Olson, A.J. Icosahedral virus structures and the protein data bank. J. Biol. Chem. 2021, 100554. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.M.; von Bonsdorff, C.H.; Nassal, M.; Fuller, S.D. Evolutionary conservation in the hepatitis B virus core structure: Comparison of human and duck cores. Structure 1995, 3, 1009–1019. [Google Scholar] [CrossRef]
- Roseman, A.M.; Berriman, J.A.; Wynne, S.A.; Butler, P.J.; Crowther, R.A. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. USA 2005, 102, 15821–15826. [Google Scholar] [CrossRef]
- Dryden, K.A.; Wieland, S.F.; Whitten-Bauer, C.; Gerin, J.L.; Chisari, F.V.; Yeager, M. Native hepatitis B virions and capsids visualized by electron cryomicroscopy. Mol. Cell 2006, 22, 843–850. [Google Scholar] [CrossRef]
- Schlicksup, C.J.; Wang, J.C.; Francis, S.; Venkatakrishnan, B.; Turner, W.W.; VanNieuwenhze, M.; Zlotnick, A. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. eLife 2018, 7. [Google Scholar] [CrossRef]
- Wu, W.; Watts, N.R.; Cheng, N.; Huang, R.; Steven, A.C.; Wingfield, P.T. Expression of quasi-equivalence and capsid dimorphism in the Hepadnaviridae. PLoS Comput. Biol. 2020, 16, e1007782. [Google Scholar] [CrossRef]
- Böttcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997, 386, 88–91. [Google Scholar] [CrossRef]
- Conway, J.F.; Cheng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94. [Google Scholar] [CrossRef]
- Böttcher, B. Hepatitis B Core Protein Capsids. Subcell. Biochem. 2021, 96, 451–470. [Google Scholar] [CrossRef]
- Watts, N.R.; Conway, J.F.; Cheng, N.; Stahl, S.J.; Belnap, D.M.; Steven, A.C.; Wingfield, P.T. The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. EMBO J. 2002, 21, 876–884. [Google Scholar] [CrossRef]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Zhang, X.; Settembre, E.; Xu, C.; Dormitzer, P.R.; Bellamy, R.; Harrison, S.C.; Grigorieff, N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. USA 2008, 105, 1867–1872. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Conley, M.J.; Bhella, D. Asymmetric analysis reveals novel virus capsid features. Biophys. Rev. 2019, 11, 603–609. [Google Scholar] [CrossRef]
- Luque, D.; Caston, J.R. Cryo-electron microscopy for the study of virus assembly. Nat. Chem. Biol. 2020, 16, 231–239. [Google Scholar] [CrossRef]
- Zlotnick, A.; Cheng, N.; Stahl, S.J.; Conway, J.F.; Steven, A.C.; Wingfield, P.T. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: Implications for morphogenesis and organization of encapsidated RNA. Proc. Natl. Acad. Sci. USA 1997, 94, 9556–9561. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.C.; Pierson, E.E.; Keifer, D.Z.; Delaleau, M.; Gallucci, L.; Cazenave, C.; Kann, M.; Jarrold, M.F.; Zlotnick, A. Importin beta Can Bind Hepatitis B Virus Core Protein and Empty Core-Like Particles and Induce Structural Changes. PLoS Pathog. 2016, 12, e1005802. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e386. [Google Scholar] [CrossRef]
- Neirynck, S.; Deroo, T.; Saelens, X.; Vanlandschoot, P.; Jou, W.M.; Fiers, W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 1999, 5, 1157–1163. [Google Scholar] [CrossRef]
- Beterams, G.; Böttcher, B.; Nassal, M. Packaging of up to 240 subunits of a 17 kDa nuclease into the interior of recombinant hepatitis B virus capsids. FEBS Lett. 2000, 481, 169–176. [Google Scholar] [CrossRef]
- Kratz, P.A.; Bottcher, B.; Nassal, M. Native display of complete foreign protein domains on the surface of hepatitis B virus capsids. Proc. Natl. Acad. Sci. USA 1999, 96, 1915–1920. [Google Scholar] [CrossRef]
- Walker, A.; Skamel, C.; Nassal, M. SplitCore: An exceptionally versatile viral nanoparticle for native whole protein display regardless of 3D structure. Sci. Rep. 2011, 1, 5. [Google Scholar] [CrossRef]
- Böttcher, B.; Vogel, M.; Ploss, M.; Nassal, M. High plasticity of the hepatitis B virus capsid revealed by conformational stress. J. Mol. Biol. 2006, 356, 812–822. [Google Scholar] [CrossRef]
- Chevreuil, M.; Lecoq, L.; Wang, S.; Gargowitsch, L.; Nhiri, N.; Jacquet, E.; Zinn, T.; Fieulaine, S.; Bressanelli, S.; Tresset, G. Nonsymmetrical Dynamics of the HBV Capsid Assembly and Disassembly Evidenced by Their Transient Species. J. Phys. Chem. B 2020, 124, 9987–9995. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, J.C.; Segura, C.P.; Hadden-Perilla, J.A.; Zlotnick, A. The Integrity of the Intradimer Interface of the Hepatitis B Virus Capsid Protein Dimer Regulates Capsid Self-Assembly. ACS Chem. Biol. 2020, 15, 3124–3132. [Google Scholar] [CrossRef]
- Lecoq, L.; Fogeron, M.L.; Meier, B.H.; Nassal, M.; Bockmann, A. Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies. Viruses 2020, 12, 1069. [Google Scholar] [CrossRef]
- Callon, M.; Malar, A.A.; Pfister, S.; Rimal, V.; Weber, M.E.; Wiegand, T.; Zehnder, J.; Chavez, M.; Cadalbert, R.; Deb, R.; et al. Biomolecular solid-state NMR spectroscopy at 1200 MHz: The gain in resolution. J. Biomol. NMR 2021. [Google Scholar] [CrossRef]
- Zlotnick, A.; Cheng, N.; Conway, J.F.; Booy, F.P.; Steven, A.C.; Stahl, S.J.; Wingfield, P.T. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 1996, 35, 7412–7421. [Google Scholar] [CrossRef]
- Vogel, M.; Diez, M.; Eisfeld, J.; Nassal, M. In vitro assembly of mosaic hepatitis B virus capsid-like particles (CLPs): Rescue into CLPs of assembly-deficient core protein fusions and FRET-suited CLPs. FEBS Lett. 2005, 579, 5211–5216. [Google Scholar] [CrossRef]
- Stray, S.J.; Johnson, J.M.; Kopek, B.G.; Zlotnick, A. An in vitro fluorescence screen to identify antivirals that disrupt hepatitis B virus capsid assembly. Nat. Biotechnol. 2006, 24, 358–362. [Google Scholar] [CrossRef]
- Ceres, P.; Zlotnick, A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 2002, 41, 11525–11531. [Google Scholar] [CrossRef]
- Lutomski, C.A.; Lyktey, N.A.; Zhao, Z.; Pierson, E.E.; Zlotnick, A.; Jarrold, M.F. Hepatitis B Virus Capsid Completion Occurs through Error Correction. J. Am. Chem. Soc. 2017, 139, 16932–16938. [Google Scholar] [CrossRef]
- Lutomski, C.A.; Lyktey, N.A.; Pierson, E.E.; Zhao, Z.; Zlotnick, A.; Jarrold, M.F. Multiple Pathways in Capsid Assembly. J. Am. Chem. Soc. 2018, 140, 5784–5790. [Google Scholar] [CrossRef]
- Hadden, J.A.; Perilla, J.R.; Schlicksup, C.J.; Venkatakrishnan, B.; Zlotnick, A.; Schulten, K. All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. eLife 2018, 7. [Google Scholar] [CrossRef]
- Perez-Segura, C.; Goh, B.C.; Hadden-Perilla, J.A. All-Atom MD Simulations of the HBV Capsid Complexed with AT130 Reveal Secondary and Tertiary Structural Changes and Mechanisms of Allostery. Viruses 2021, 13, 564. [Google Scholar] [CrossRef]
- Patterson, A.; Zhao, Z.; Waymire, E.; Zlotnick, A.; Bothner, B. Dynamics of Hepatitis B Virus Capsid Protein Dimer Regulate Assembly through an Allosteric Network. ACS Chem. Biol. 2020, 15, 2273–2280. [Google Scholar] [CrossRef]
- Makbul, C.; Khayenko, V.; Maric, H.M.; Bottcher, B. Conformational Plasticity of Hepatitis B Core Protein Spikes Promotes Peptide Binding Independent of the Secretion Phenotype. Microorganisms 2021, 9, 956. [Google Scholar] [CrossRef]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef]
- Zlotnick, A.; Johnson, J.M.; Wingfield, P.W.; Stahl, S.J.; Endres, D. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 1999, 38, 14644–14652. [Google Scholar] [CrossRef]
- Singh, S.; Zlotnick, A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J. Biol. Chem. 2003, 278, 18249–18255. [Google Scholar] [CrossRef]
- Spiriti, J.; Conway, J.F.; Zuckerman, D.M. Should Virus Capsids Assemble Perfectly? Theory and Observation of Defects. Biophys. J. 2020, 119, 1781–1790. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, J.C.; Zhang, M.; Lyktey, N.A.; Jarrold, M.F.; Jacobson, S.C.; Zlotnick, A. Asymmetrizing an icosahedral virus capsid by hierarchical assembly of subunits with designed asymmetry. Nat. Commun. 2021, 12, 589. [Google Scholar] [CrossRef]
- Newman, M.; Chua, P.K.; Tang, F.M.; Su, P.Y.; Shih, C. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J. Virol. 2009, 83, 10616–10626. [Google Scholar] [CrossRef]
- He, L.; Porterfield, Z.; van der Schoot, P.; Zlotnick, A.; Dragnea, B. Hepatitis virus capsid polymorph stability depends on encapsulated cargo size. ACS Nano 2013, 7, 8447–8454. [Google Scholar] [CrossRef]
- Dhason, M.S.; Wang, J.C.; Hagan, M.F.; Zlotnick, A. Differential assembly of Hepatitis B Virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded. Virology 2012, 430, 20–29. [Google Scholar] [CrossRef]
- Petrovskis, I.; Lieknina, I.; Dislers, A.; Jansons, J.; Bogans, J.; Akopjana, I.; Zakova, J.; Sominskaya, I. Production of the HBc Protein from Different HBV Genotypes in E. coli. Use of Reassociated HBc VLPs for Packaging of ss- and dsRNA. Microorganisms 2021, 9, 283. [Google Scholar] [CrossRef]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Twarock, R.; Stockley, P.G. RNA-Mediated Virus Assembly: Mechanisms and Consequences for Viral Evolution and Therapy. Annu. Rev. Biophys. 2019, 48, 495–514. [Google Scholar] [CrossRef]
- Comas-Garcia, M. Packaging of Genomic RNA in Positive-Sense Single-Stranded RNA Viruses: A Complex Story. Viruses 2019, 11, 253. [Google Scholar] [CrossRef]
- Patel, N.; White, S.J.; Thompson, R.F.; Bingham, R.; Weiss, E.U.; Maskell, D.P.; Zlotnick, A.; Dykeman, E.; Tuma, R.; Twarock, R.; et al. HBV RNA pre-genome encodes specific motifs that mediate interactions with the viral core protein that promote nucleocapsid assembly. Nat. Microbiol. 2017, 2, 17098. [Google Scholar] [CrossRef]
- Oliver, R.C.; Potrzebowski, W.; Najibi, S.M.; Pedersen, M.N.; Arleth, L.; Mahmoudi, N.; Andre, I. Assembly of Capsids from Hepatitis B Virus Core Protein Progresses through Highly Populated Intermediates in the Presence and Absence of RNA. ACS Nano 2020, 14, 10226–10238. [Google Scholar] [CrossRef]
- Junker-Niepmann, M.; Bartenschlager, R.; Schaller, H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990, 9, 3389–3396. [Google Scholar] [CrossRef]
- Knaus, T.; Nassal, M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993, 21, 3967–3975. [Google Scholar] [CrossRef]
- Anasir, M.I.; Zarif, F.; Poh, C.L. Antivirals blocking entry of enteroviruses and therapeutic potential. J. Biomed. Sci. 2021, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N. HIV Capsid and Integration Targeting. Viruses 2021, 13, 125. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Quinn, C.M.; Perilla, J.R.; Zhang, H.; Shirra, R., Jr.; Hou, G.; Byeon, I.J.; Suiter, C.L.; Ablan, S.; Urano, E.; et al. Quenching protein dynamics interferes with HIV capsid maturation. Nat. Commun. 2017, 8, 1779. [Google Scholar] [CrossRef]
- King, R.W.; Ladner, S.K.; Miller, T.J.; Zaifert, K.; Perni, R.B.; Conway, S.C.; Otto, M.J. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-L-2’,3’-dideoxy-3’-thiacytidine. Antimicrob. Agents Chemother. 1998, 42, 3179–3186. [Google Scholar] [CrossRef]
- Delaney, W.E.t.; Edwards, R.; Colledge, D.; Shaw, T.; Furman, P.; Painter, G.; Locarnini, S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3057–3060. [Google Scholar] [CrossRef]
- Weber, O.; Schlemmer, K.H.; Hartmann, E.; Hagelschuer, I.; Paessens, A.; Graef, E.; Deres, K.; Goldmann, S.; Niewoehner, U.; Stoltefuss, J.; et al. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antivir. Res. 2002, 54, 69–78. [Google Scholar] [CrossRef]
- Deres, K.; Schroder, C.H.; Paessens, A.; Goldmann, S.; Hacker, H.J.; Weber, O.; Kramer, T.; Niewohner, U.; Pleiss, U.; Stoltefuss, J.; et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [Google Scholar] [CrossRef]
- Campagna, M.R.; Liu, F.; Mao, R.; Mills, C.; Cai, D.; Guo, F.; Zhao, X.; Ye, H.; Cuconati, A.; Guo, H.; et al. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J. Virol. 2013, 87, 6931–6942. [Google Scholar] [CrossRef]
- Nijampatnam, B.; Liotta, D.C. Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [Google Scholar] [CrossRef]
- Zlotnick, A.; Lee, A.; Bourne, C.R.; Johnson, J.M.; Domanico, P.L.; Stray, S.J. In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions). Nat. Protoc. 2007, 2, 490–498. [Google Scholar] [CrossRef]
- Senaweera, S.; Du, H.; Zhang, H.; Kirby, K.A.; Tedbury, P.R.; Xie, J.; Sarafianos, S.G.; Wang, Z. Discovery of New Small Molecule Hits as Hepatitis B Virus Capsid Assembly Modulators: Structure and Pharmacophore-Based Approaches. Viruses 2021, 13, 770. [Google Scholar] [CrossRef]
- Katen, S.P.; Chirapu, S.R.; Finn, M.G.; Zlotnick, A. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol. 2010, 5, 1125–1136. [Google Scholar] [CrossRef]
- Stray, S.J.; Bourne, C.R.; Punna, S.; Lewis, W.G.; Finn, M.G.; Zlotnick, A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Lahlali, T.; Berke, J.M.; Vergauwen, K.; Foca, A.; Vandyck, K.; Pauwels, F.; Zoulim, F.; Durantel, D. Novel Potent Capsid Assembly Modulators Regulate Multiple Steps of the Hepatitis B Virus Life Cycle. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Kondylis, P.; Schlicksup, C.J.; Katen, S.P.; Lee, L.S.; Zlotnick, A.; Jacobson, S.C. Evolution of Intermediates during Capsid Assembly of Hepatitis B Virus with Phenylpropenamide-Based Antivirals. ACS Infect. Dis. 2019, 5, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Bourne, C.R.; Finn, M.G.; Zlotnick, A. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J. Virol. 2006, 80, 11055–11061. [Google Scholar] [CrossRef]
- Packianathan, C.; Katen, S.P.; Dann, C.E., III; Zlotnick, A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J. Virol. 2010, 84, 1607–1615. [Google Scholar] [CrossRef]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. USA 2015, 112, 15196–15201. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci. Rep. 2017, 7, 42374. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Cheng, J.; Hu, Z.; Ban, H.; Wu, S.; Hwang, N.; Kulp, J.; Li, Y.; Du, Y.; Chang, J.; et al. Identification of hepatitis B virus core protein residues critical for capsid assembly, pgRNA encapsidation and resistance to capsid assembly modulators. Antivir. Res. 2021, 191, 105080. [Google Scholar] [CrossRef]
- Venkatakrishnan, B.; Katen, S.P.; Francis, S.; Chirapu, S.; Finn, M.G.; Zlotnick, A. Hepatitis B Virus Capsids Have Diverse Structural Responses to Small-Molecule Ligands Bound to the Heteroaryldihydropyrimidine Pocket. J. Virol. 2016, 90, 3994–4004. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fan, G.; Wang, Z.; Chen, H.S.; Yin, C.C. Allosteric conformational changes of human HBV core protein transform its assembly. Sci. Rep. 2017, 7, 1404. [Google Scholar] [CrossRef]
- Huang, Q.; Cai, D.; Yan, R.; Li, L.; Zong, Y.; Guo, L.; Mercier, A.; Zhou, Y.; Tang, A.; Henne, K.; et al. Preclinical Profile and Characterization of the Hepatitis B Virus Core Protein Inhibitor ABI-H0731. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Schlicksup, C.J.; Laughlin, P.; Dunkelbarger, S.; Wang, J.C.; Zlotnick, A. Local Stabilization of Subunit-Subunit Contacts Causes Global Destabilization of Hepatitis B Virus Capsids. ACS Chem. Biol. 2020, 15, 1708–1717. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, J.; Tang, L.; Hu, Z.; Luo, Y.; Li, Y.; Zhou, T.; Chang, J.; Guo, J.T. Virological Basis for the Cure of Chronic Hepatitis B. ACS Infect. Dis. 2019, 5, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef]
- Kayesh, M.E.H.; Sanada, T.; Kohara, M.; Tsukiyama-Kohara, K. Tree Shrew as an Emerging Small Animal Model for Human Viral Infection: A Recent Overview. Viruses 2021, 13, 1641. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Strick-Marchand, H. In-vitro and in-vivo models for hepatitis B cure research. Curr. Opin. HIV AIDS 2020, 15, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Maya, S.; Ploss, A. Animal Models of Hepatitis B Virus Infection-Success, Challenges, and Future Directions. Viruses 2021, 13, 777. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wu, M.; Ghildyal, R.; Yuan, Z. Animal Models for the Study of Hepatitis B Virus Pathobiology and Immunity: Past, Present, and Future. Front. Microbiol. 2021, 12, 715450. [Google Scholar] [CrossRef]
- Acs, G.; Sells, M.A.; Purcell, R.H.; Price, P.; Engle, R.; Shapiro, M.; Popper, H. Hepatitis B virus produced by transfected Hep G2 cells causes hepatitis in chimpanzees. Proc. Natl. Acad. Sci. USA 1987, 84, 4641–4644. [Google Scholar] [CrossRef]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef]
- Sun, D.; Nassal, M. Stable HepG2- and Huh7-based human hepatoma cell lines for efficient regulated expression of infectious hepatitis B virus. J. Hepatol. 2006, 45, 636–645. [Google Scholar] [CrossRef]
- Schultz, U.; Grgacic, E.; Nassal, M. Duck hepatitis B virus: An invaluable model system for HBV infection. Adv. Virus. Res. 2004, 63, 1–70. [Google Scholar] [CrossRef]
- Roggendorf, M.; Kosinska, A.D.; Liu, J.; Lu, M. The Woodchuck, a Nonprimate Model for Immunopathogenesis and Therapeutic Immunomodulation in Chronic Hepatitis B Virus Infection. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Rat, V.; Seigneuret, F.; Burlaud-Gaillard, J.; Lemoine, R.; Hourioux, C.; Zoulim, F.; Testoni, B.; Meunier, J.C.; Tauber, C.; Roingeard, P.; et al. BAY 41-4109-mediated aggregation of assembled and misassembled HBV capsids in cells revealed by electron microscopy. Antivir. Res. 2019, 169, 104557. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Core autophagy genes and human diseases. Curr. Opin. Cell Biol. 2019, 61, 117–125. [Google Scholar] [CrossRef]
- Tan, Z.; Pionek, K.; Unchwaniwala, N.; Maguire, M.L.; Loeb, D.D.; Zlotnick, A. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J. Virol. 2015, 89, 3275–3284. [Google Scholar] [CrossRef]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Mostmans, W.; Vandyck, K.; Raboisson, P.; Pauwels, F. Antiviral Properties and Mechanism of Action Studies of the Hepatitis B Virus Capsid Assembly Modulator JNJ-56136379. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Bester, R.; Zhou, X.; Xu, Z.; Blossey, C.; Sacherl, J.; Vondran, F.W.R.; Gao, L.; Protzer, U. A New Role for Capsid Assembly Modulators to Target Mature Hepatitis B Virus Capsids and Prevent Virus Infection. Antimicrob. Agents Chemother. 2019, 64. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Ren, S.; Espiritu, C.; Kelly, M.; Lau, V.; Zheng, L.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Hepatitis B Virus Capsid Assembly Modulators, but Not Nucleoside Analogs, Inhibit the Production of Extracellular Pregenomic RNA and Spliced RNA Variants. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Klumpp, K.; Shimada, T.; Allweiss, L.; Volz, T.; Lutgehetmann, M.; Hartman, G.; Flores, O.A.; Lam, A.M.; Dandri, M. Efficacy of NVR 3-778, Alone and In Combination with Pegylated Interferon, vs. Entecavir In uPA/SCID Mice With Humanized Livers and HBV Infection. Gastroenterology 2018, 154, 652–662.e658. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.S.C.; Walker, K.; Flores, O.A.; Hartmann, G.D.; Klumpp, K.; Liaw, S.; Brown, N.A. Phase 1a safety and pharmacokinetics of NVR 3-778, a potential first-in-class HBV core inhibitor. Hepatology 2014, 60, 1267A–1290A. [Google Scholar]
- Lam, A.M.; Espiritu, C.; Vogel, R.; Ren, S.; Lau, V.; Kelly, M.; Kuduk, S.D.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e1397. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Lenz, O.; Vandenbossche, J.J.; Talloen, W.; Verbinnen, T.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; Crespo, J.; et al. JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and Has Antiviral Activity in a Phase 1 Study of Patients with Chronic Infection. Gastroenterology 2020, 159, 521–533.e529. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.; Hou, J.; Asselah, T.; Chan, H.; Zoulim, F.; Tanaka, Y.; Janczewska, E.; Nahass, R.; Bourgeois, S.; Buti, M.; et al. Efficacy and safety results of the phase 2 JNJ-56136379 JADE study in patients with chronic hepatitis B: Interim week 24 data. J. Hepatol. 2020, 73, S129–S130. [Google Scholar] [CrossRef]
- Gane, E.; Schwabe, C.; Lenz, O.; Verbinnen, T.; Talloen, W.; Kakuda, T.N.; Westland, C.; Patel, M.; Yogaratnam, J.; Dragone, L.; et al. JNJ-64530440 (JNJ-0440), A Novel, Class N Capsid Assembly Modulator (CAM-N): Safety, Tolerability, Pharmacokinetics and Antiviral Activity of Multiple Ascending Doses in Patients with Chronic Hepatitis B. Liver Meet. Boston AASLD 2019. [Google Scholar]
- Yuen, M.F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166. [Google Scholar] [CrossRef]
- Agarwal, K.; Niu, J.; Ding, Y.; Gane, E.; Nguyen, T.; Alves, K.; Evanchick, M.; Zayed, H.; Huang, Q.; Knox, S.; et al. Antiviral activity, pharmacokinetics and safety of the second- generation hepatitis B core inhibitor ABI-H2158 in Phase 1b study of patients with HBeAg-positive chronic hepatitis B infection. J. Hepatol. 2020, 73, S123–S400. [Google Scholar] [CrossRef]
- Huang, Q.; Haydar, S.; Zhou, Y.; Cai, D.; Xu, X.; Yan, R.; Carabajal, E.; Tang, X.; Walker, A.M.; Rai, R.; et al. Preclinical Profile of HBV Core Protein Inhibitor, ABI-H3733, a Potent Inhibitor of cccDNA Generation in HBV Infected Cells. J. Hepatol. 2019, 70, e1–e952. [Google Scholar]
- Feng, S.; Gane, E.; Schwabe, C.; Zhu, M.; Triyatni, M.; Zhou, J.; Bo, Q.; Jin, Y. A Five-in-One First-in-Human Study To Assess Safety, Tolerability, and Pharmacokinetics of RO7049389, an Inhibitor of Hepatitis B Virus Capsid Assembly, after Single and Multiple Ascending Doses in Healthy Participants. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Zhou, X.; Gane, E.; Schwabe, C.; Tanwandee, T.; Feng, S.; Jin, Y.; Triyatni, M.; Lemenuel-Diot, A.; Cosson, V.; et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: A multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 723–732. [Google Scholar] [CrossRef]
- Yuen, M.F.; Berliba, E.; Sukeepaisarnjaroen, W.; Ahn, S.H.; Tanwandee, T.; Lim, Y.S.; Kim, Y.J.; Poovarawan, K.; Tangkijvanich, O.; Chan, H.L.Y.; et al. Safety, Tolerability, Pharmacokinetics (PK), and Antiviral Activity of the Capsid Inhibitor (CI) AB-506 in Healthy Subjects (HS) and Chronic Hepatitis B (CHB) Subjects. Hepatology 2019, 70, 1477A–1501A. [Google Scholar] [CrossRef]
- Mani, N.; Cole, A.; Kultgen, S.; Ardzinski, A.; Chiu, T.; Cuconati, A.; Dorsey, B.D.; Fan, K.; Graves, I.; Guo, J.T.; et al. Preclinical antiviral profile of AB-836, a potent, highly selective hepatitis B virus capsid inhibitor. J. Hepatol. 2021, 75, S201–S293. [Google Scholar]
- Vaine, M.; Dellisola, V.; Clugston, S.; Cao, H.; Gao, X.; Kass, J.; Li, W.; Peng, X.; Qiu, Y.-L.; Jiang, L.; et al. FRI-191-EDP-514, a novel HBV core inhibitor with potent antiviral activity both in vitro and in vivo. J. Hepatol. 2019, 70, e474–e475. [Google Scholar] [CrossRef]
- Li, C.; Wu, M.; Zhang, H.; Mai, J.; Yang, L.; Ding, Y.; Niu, J.; Mao, J.; Wu, W.; Zhang, D.; et al. Safety, tolerability, and pharmacokinetics of the novel hepatitis B virus capsid assembly modulator GST-HG141 in healthy Chinese subjects: A first-in-human single- and multiple-dose escalation trial. Antimicrob. Agents Chemother. 2021, 65, e:0122021. [Google Scholar] [CrossRef]
- Coburn, G.; Benetatos, C.; Yao, J.; Boyd, S.; Haimowitz, T.; Condon, S.; Drager, T.; Hart, S.E.; Pevear, D. Discovery and preclinical profile of VNRX-9945, a potent, broadly active core protein inhibitor for the treatment of hepatitis B virus (HBV) infection. J. Hepatol. 2021, 75, S294–S803. [Google Scholar]
- Zhang, Q.; Vendeville, S.; Serebryany, V.; Welch, M.; Liu, J.; Williams, C.; Debing, Y.; Jekle, A.; Stevens, S.; Deval, J.; et al. ALG-000184, a prodrug of capsid assembly modulator ALG-001075, demonstrates best-in-class preclinical characteristics for the treatment of chronic hepatitis B. J. Hepatol. 2020, 73, S880–S881. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Tan, Y.; Xin, Y.; Gao, H.; Zheng, S.; Yi, Y.; Zhang, J.; Wu, C.; Zhao, Y.; et al. Efficacy and safety of GLS4/ritonavir combined with entecavir in HBeAg-positive patients with chronic hepatitis B: Interim results from phase 2b, multi-center study. J. Hepatol. 2020, 73, S878–S880. [Google Scholar] [CrossRef]
- Tai, Z.; Tian, Q.; Zhao, X.; Xie, J.; Lu, Y.; Tan, Y.; Zhao, W.; Ma, X.; Yuan, X.; Song, H.; et al. Discovery of KL060332, a potential best-in-class capsid inhibitor. Hepatology 2020, 72, 504A. [Google Scholar]
- Debing, Y.; Kum, D.B.; Liu, C.; Deval, J.; Vanrusselt, H.; Sanchez, A.A.; Zhang, Q.; Mukherjee, S.; Misner, D.; Chanda, S.; et al. Capsid assembly modulator ALG-000111 and its prodrug ALG-000286 display excellent in vitro and in vivo antiviral activity. J. Hepatol. 2021, 75, S294–S803. [Google Scholar]
- Amblard, F.; Boucle, S.; Bassit, L.; Cox, B.; Sari, O.; Tao, S.; Chen, Z.; Ozturk, T.; Verma, K.; Russell, O.; et al. Novel Hepatitis B Virus Capsid Assembly Modulator Induces Potent Antiviral Responses In Vitro and in Humanized Mice. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Di Fabio, R.; Bencheva, L.; De Francesco, R.; Donnici, L.; Summa, V.; Monteagudo, E.; Iannacone, M.; Guidotti, L.G. Preclinical assessment of capsid assembly modulators (CAMs) of varying potency revealed a novel class of picomolar-acting CAMs inducing neither significant HBcAg cytoplasmic retention nor adverse interactions with HBcAg-specific adaptive immunity. J. Hepatol. 2020, 73, S5. [Google Scholar] [CrossRef]
- Verbinnen, T.; Hodari, M.; Talloen, W.; Berke, J.M.; Blue, D.; Yogaratnam, J.; Vandenbossche, J.; Shukla, U.; De Meyer, S.; Lenz, O. Virology analysis of chronic hepatitis B virus-infected patients treated for 28 days with JNJ-56136379 monotherapy. J. Viral Hepat. 2020, 27, 1127–1137. [Google Scholar] [CrossRef]
- Verbinnen, T.; Talloen, W.; Shukla, U.; Vandenbossche, J.; Biermer, M.; Beumont-Mauviel, M.; De Meyer, S.; Lenz, O. Viral sequence analysis of chronic hepatitis B (CHB) patients treated with the capsid assembly modulator (CAM-N) JNJ-56136379 (JNJ-6379) as monotherapy in the ongoing JADE phase 2a study. (POSTER 856). Hepatology 2020, 72, 131A–1159A. [Google Scholar] [CrossRef]
- Mak, L.Y.; Seto, W.K.; Yuen, M.F. Novel Antivirals in Clinical Development for Chronic Hepatitis B Infection. Viruses 2021, 13, 1169. [Google Scholar] [CrossRef]
- Gane, E.; Sulkowski, M.; Ma, X.; Nguyen, T.; Hann, H.; Hassanein, T.; Elkhashab, M.; Nahass, R.; Chan, S.; Bennet, M.; et al. Viral response and safety following discontinuation of treatment with the core inhibitor vebicorvir and a nucleos(t)ide reverse transcriptase inhibitor in patients with HBeAg positive or negative chronic hepatitis B virus infection. J. Hepatol. 2021, in press. [Google Scholar]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Laskey, S.B.; Siliciano, R.F. A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat. Rev. Microbiol. 2014, 12, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Dousson, C.B.; Lampertico, P. Is there any need for new, long-acting nucleos(t)ide analogues for the treatment of hepatitis B infection? J. Hepatol. 2021, 74, 1011–1014. [Google Scholar] [CrossRef]
- Higashi-Kuwata, N.; Hayashi, S.; Kumamoto, H.; Ogata-Aoki, H.; Das, D.; Venzon, D.; Hattori, S.I.; Bulut, H.; Hashimoto, M.; Otagiri, M.; et al. Identification of a novel long-acting 4’-modified nucleoside reverse transcriptase inhibitor against HBV. J. Hepatol. 2021, 74, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, E.; Suwanmanee, Y.; Uede, K. Chronic Hepatitis B Treatment Strategies using Polymerase Inhibitor-based Combination Therapy. Viruses 2021, 13, 1691. [Google Scholar] [CrossRef]
- Pierra Rouviere, C.; Dousson, C.B.; Tavis, J.E. HBV replication inhibitors. Antivir. Res. 2020, 179, 104815. [Google Scholar] [CrossRef] [PubMed]
- Nishio, A.; Bolte, F.J.; Takeda, K.; Park, N.; Yu, Z.X.; Park, H.; Valdez, K.; Ghany, M.G.; Rehermann, B. Clearance of pegylated interferon by Kupffer cells limits NK cell activation and therapy response of patients with HBV infection. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Yuen, M.F.; Locarnini, S.; Given, B.; Schluep, T.; Hamilton, J.; Biermer, M.; Kalmeijer, R.; Beumont-Mauviel, M.; Lenz, O.; Cloherty, G.; et al. First Clinical Experience with RNA Interference (RNAi)-based Triple Combination Therapy in Chronic Hepatitis B (CHB): JNJ-73763989 (JNJ-3989), JNJ-56136379 (JNJ-6379) and a Nucleos(t)ide Analogue (NA). Hepatology 2019, 70, 1489A. [Google Scholar]
- Berke, J.M.; Tan, Y.; Verbinnen, T.; Dehertogh, P.; Vergauwen, K.; Vos, A.; Lenz, O.; Pauwels, F. Antiviral profiling of the capsid assembly modulator BAY41-4109 on full-length HBV genotype A-H clinical isolates and core site-directed mutants in vitro. Antivir. Res. 2017, 144, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chang, S.; Hsieh, D.; Burdette, D.; Martin, R.; Mo, H.; Feierbach, B. Generation of an HBV core phenotyping assay for evaluating HBV capsid compounds. J. Virol. Methods 2021, 292, 114117. [Google Scholar] [CrossRef]
- Wu, S.; Luo, Y.; Viswanathan, U.; Kulp, J.; Cheng, J.; Hu, Z.; Xu, Q.; Zhou, Y.; Gong, G.Z.; Chang, J.; et al. CpAMs induce assembly of HBV capsids with altered electrophoresis mobility: Implications for mechanism of inhibiting pgRNA packaging. Antivir. Res. 2018, 159, 1–12. [Google Scholar] [CrossRef]
- Lee, A.C.H.; Thi, E.P.; Ardzinski, A.; Brown, J.; Eley, T.; Mani, N.; Rijmbrand, R.; Sims, K.; Sofia, M.J.; Picchio, G. Hepatitis B virus core protein variants observed in a first-in-human placebo-controlled study of a core protein inhibitor. J. Hepatol. 2020, 73, S833. [Google Scholar] [CrossRef]
- Yuen, M.F.; Locarnini, S.; Revill, P.A.; Yan, R.; Ouyang, L.; Cai, D.; Delaney, W.; Kitrinos, K.M.; Thompson, A.; Zoulim, F.; et al. No emergent core inhibitor resistance in patients with chronic hepatitis B virus infection treated with Vebicorvir in combination with a nucleos (t)ide reverse transcriptase inhibitor. J. Hepatol. 2021, 75, S294–S803. [Google Scholar]
- Kim, C.; Barnes, L.; Schlicksup, C.J.; Patterson, A.; Bothner, B.; Jarrold, M.; Wang, C.J.; Zlotnick, A. Core Protein-Directed Antivirals and Importin b Can Synergistically Disrupt HBV Capsids. bioRxiv Prepr. 2021. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAM/CAM Type | Chemo- Type | Phase | In Vitro EC50 [nM] a | Mean log10 Reduction at End of Treatment Period (TP) | Trial ID c | Combination Trial Agents d | Sponsor | Refs. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| DNAic/DNAec | RNAic-t/pgRNAic/HBs | cccDNA | DNA/RNA (TP) b | |||||||
| Prototypical CAMs | ||||||||||
| AT-130/E | PPA | P/C | ~200–2500/ND | ND/ND/ND | ND | NA | NA | [185] | ||
| DVR-23/E | SBA | P/C | 779/ND | ND/ND/ND | ND | NA | NA | [188] | ||
| BAY 41-4109/A | HAP | 1; DC | 33–276/ND | ND/ND/ND | ND | ND | ND | NA | AiCuris | [186] |
| CAMs in recent and ongoing trials | ||||||||||
| NVR 3-778/E | SBA | 1; DC | 340/440 | 3700/ND/4800 | ND | 1.43/1.42 (28 d) | NCT02401737 | pIFN | Novira/J&J | [226,227] |
| JNJ-6379/E | SPA | 2a/b | 54–69/102 | 876/ND/1608 | ND | 2.70/1.83 (28 d) | NCT04129554, NCT03982186, NCT04667104, NCT04439539 | NUC NUC + pIFN NUC + pIFN + JNJ-3989 (a-HBV siRNA) | Janssen/J&J | [220,228,229] |
| JNJ-0440/E | SBA | 1b | 12–24/NDR | 136/ND/243 | ND | 3.3/2.6 (28 d) | NCT03439488 | NA | Janssen/J&J | [230] |
| ABI-H0731 (Vebi- corvir)/E | DBT | 2a; DC | 154–307/ND | ND/~2500/~5000 | ND | 2.8/2.0 (28 d) | NCT03109730, NCT03576066, NCT03577171, NCT04781647, NCT04820686 | NUC NUC + pIFN NUC + AB-729 (a-HBV siRNA) | Assembly Biosciences | [203,231] |
| ABI-H2158/E | ND | 2a; DC | 22/ND | ND/227/ND | 334 | 2.5/2.2 (14 d) | NCT04398134 | NUC | Assembly Biosciences | [232] |
| ABI-H3733/E | ND | 1b | 5–12/ND | ND/27–80/43–77 | 125 | TBR | NCT04271592 | NA | Assembly Biosciences | [233] |
| RO7049389/A | HAP | 2a | 4–62/6.0 | ND/ND/~1000 | ND | 2.86/2.54 (28 d) | NCT03570658, NCT03717064, NCT02952924, NCT04225715 | NUC + RO7020531 (TLR7 agonist) NUC + RO7445482 (a-HBV siRNA) | Hoffmann-LaRoche | [234,235] |
| AB-506/E | Amino-indane | 1; DC | 65–77/ND | ND/ND/1430 | ND | ~2.2/~2.5 (28 d) | NDR | NA | Arbutus | [236] |
| AB-836/E | ND | 1a/b | 2–12/ND | ND/ND/197 | 175 | TBR | NCT04775797 | NA | Arbutus | [237] |
| EDP-514/E | ND | 1b | 17–27/ND | ND/3–25/35 | ND | 3.3/2.4 (28 d) | NCT04008004, NCT04470388, NCT04971512 | EDP-721 (HBV RNA destabilizer) | Enanta | [238] |
| GST-HG141/E | ND | 1a/b | ND/8/ND | ND/ND/ND | ND | TBR | NCT04386915, NCT04868981 | NA | Fujian Cosunter | [239] |
| VNRX-9945/E | ND | 1a | ND/2.3–10 | ND/ND/90 | ND | NA | NCT04845321 | NA | Venatorx | [240] |
| ALG-000184 (prodrug of ALG-001075)/E | ND | 1b | 0.5–1.4/ND | 54/ND/70 | 2.9/ND (14 d) | NCT04536337 | ALG-010133 (HBsAg release inhibitor), ALG-125755 (siRNA), ALG-020572 (ASO), NUC | Aligos | [241] | |
| QL-007/ND | ND | 2a | ND | ND | ND | ND | NCT04157699, NCT04157257 | NUC | Qilu | see NCT |
| ZM-H1505R/ND | Pyrazole | 1a | ND | ND | ND | NA | NCT04220801 | NA | Shanghai Zhimeng | see NCT |
| GLS4/RTV (CYP block)/A | HAP | 2a | ND | ND | ND | 4.37/2.47 (24 week) | NCT04147208 | NUC | Sunshine Lake | [242] |
| KL060332/A | HAP | 1a | 5.0 (n.s.) | ND | ND | NDR | CTR20200985 | NA | Kelun-Biotech | [243] |
| New CAMs in current preclinical trials | ||||||||||
| ABI-H4334/ND | ND | P/C | 0.5/2.4 (n.s.) | ND | ND | NA | NA | NA | Assembly Biosciences | e |
| ALG-000286 (prodrug of ALG-000111)/E | ND | P/C | 0.7–0.9/ND | 34/ND/42 | ND | NA | NA | NA | Aligos | [244] |
| GLP-26/E | Glyoxamido-pyrrole | P/C | ND/3–4 | ND/11/ND | ND | NA | NA | NA | Emory University | [245] |
| M-1428/E | ND | P/C | “500-fold higher potency vs. NVR 3-778” (n.s.) | ND/ND/ND | ND | NA | NA | NA | Vita-Salute San Raffaele University | [246] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niklasch, M.; Zimmermann, P.; Nassal, M. The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target. Biomedicines 2021, 9, 1577. https://doi.org/10.3390/biomedicines9111577
Niklasch M, Zimmermann P, Nassal M. The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target. Biomedicines. 2021; 9(11):1577. https://doi.org/10.3390/biomedicines9111577
Chicago/Turabian StyleNiklasch, Matthias, Peter Zimmermann, and Michael Nassal. 2021. "The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target" Biomedicines 9, no. 11: 1577. https://doi.org/10.3390/biomedicines9111577
APA StyleNiklasch, M., Zimmermann, P., & Nassal, M. (2021). The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target. Biomedicines, 9(11), 1577. https://doi.org/10.3390/biomedicines9111577