
Mitoxantrone-Loaded Nanoferritin Slows Tumor Growth and Improves the Overall Survival Rate in a Subcutaneous Pancreatic Cancer Mouse Model

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. HFt-MP-PASE-MIT Production
2.2. The In Vitro Viability XTT Assays and Kinetic Cell Intoxication
2.3. Apoptosis Evaluation by Flow Cytometry
2.4. Drug Uptake by Tumor Cells
2.5. In Vivo Therapeutic Evaluation
2.6. Histological Evaluation of Primary Tumors and Other Organs
2.7. Statistical Analysis
3. Results
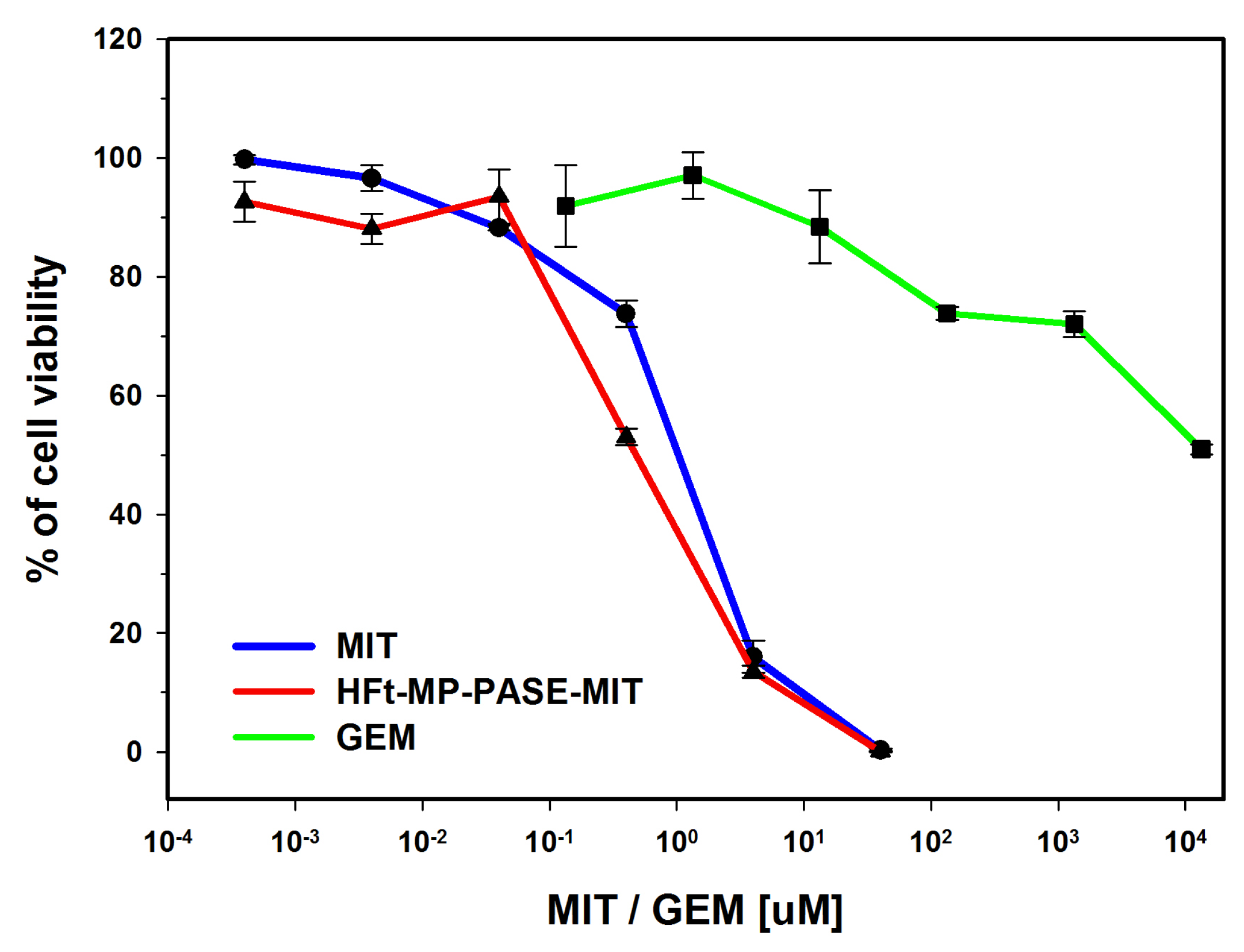
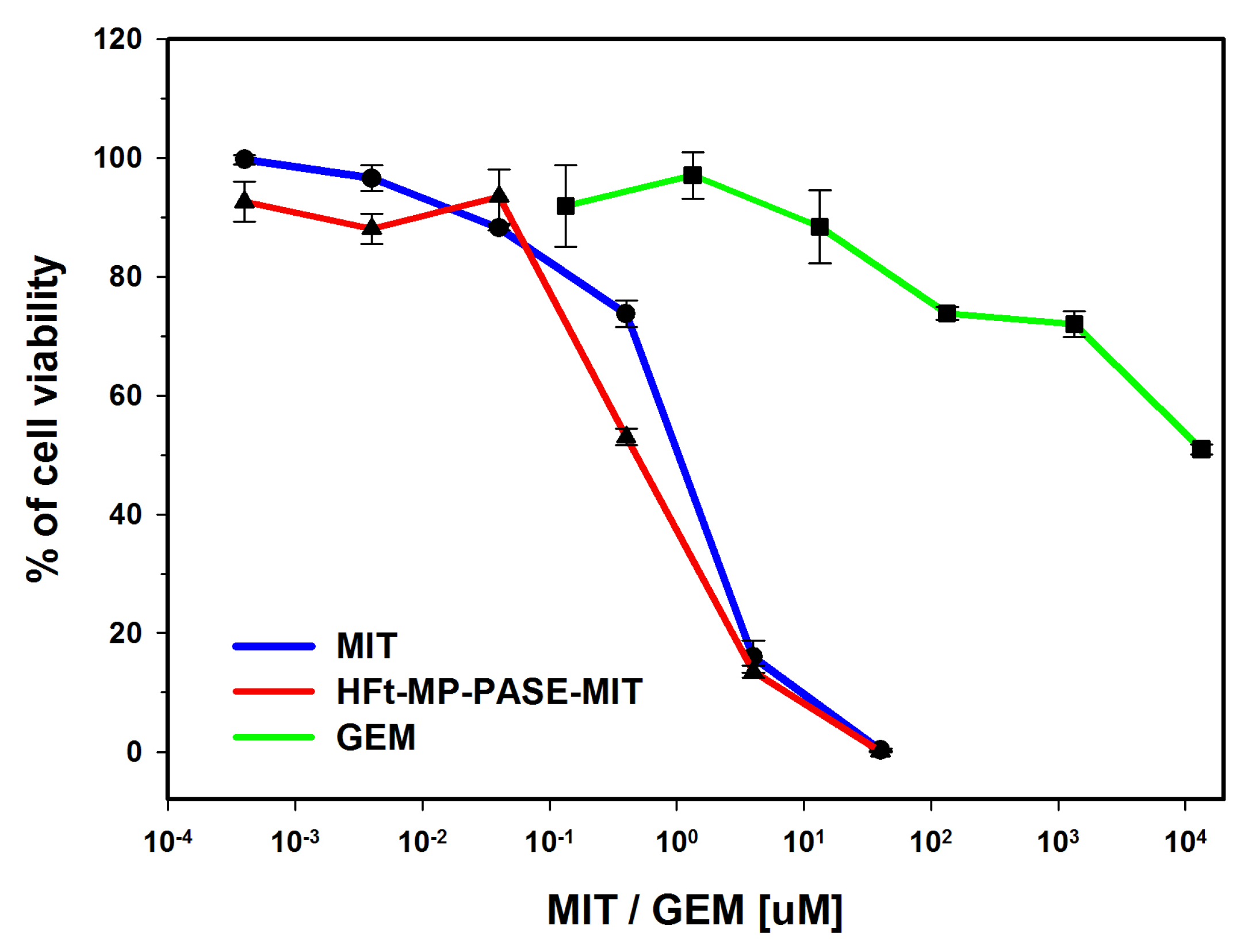
3.1. In Vitro Cytotoxicity on PaCa44 Cells
3.2. Flow Cytometric Analysis of Necrosis or Apoptosis
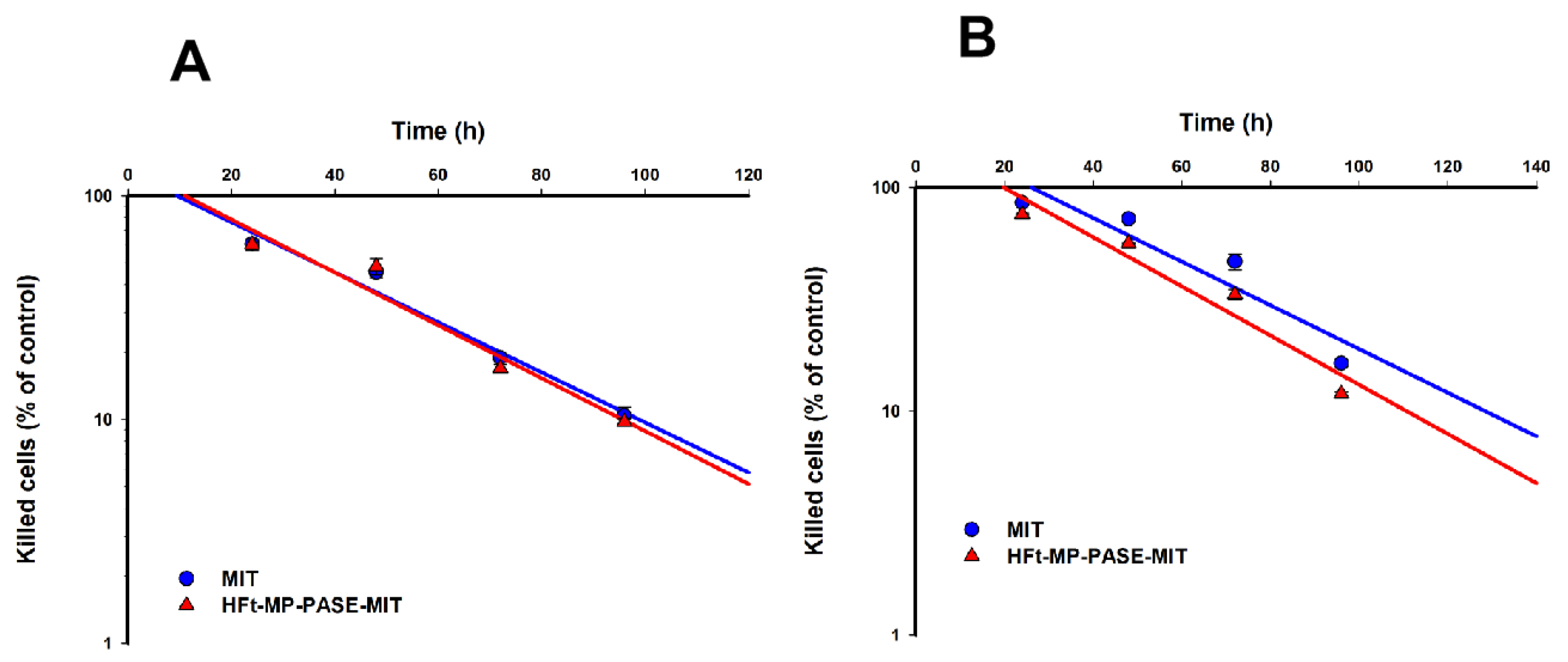
3.3. Kinetics of Cell Intoxication
3.4. Uptake in Tumor Cells
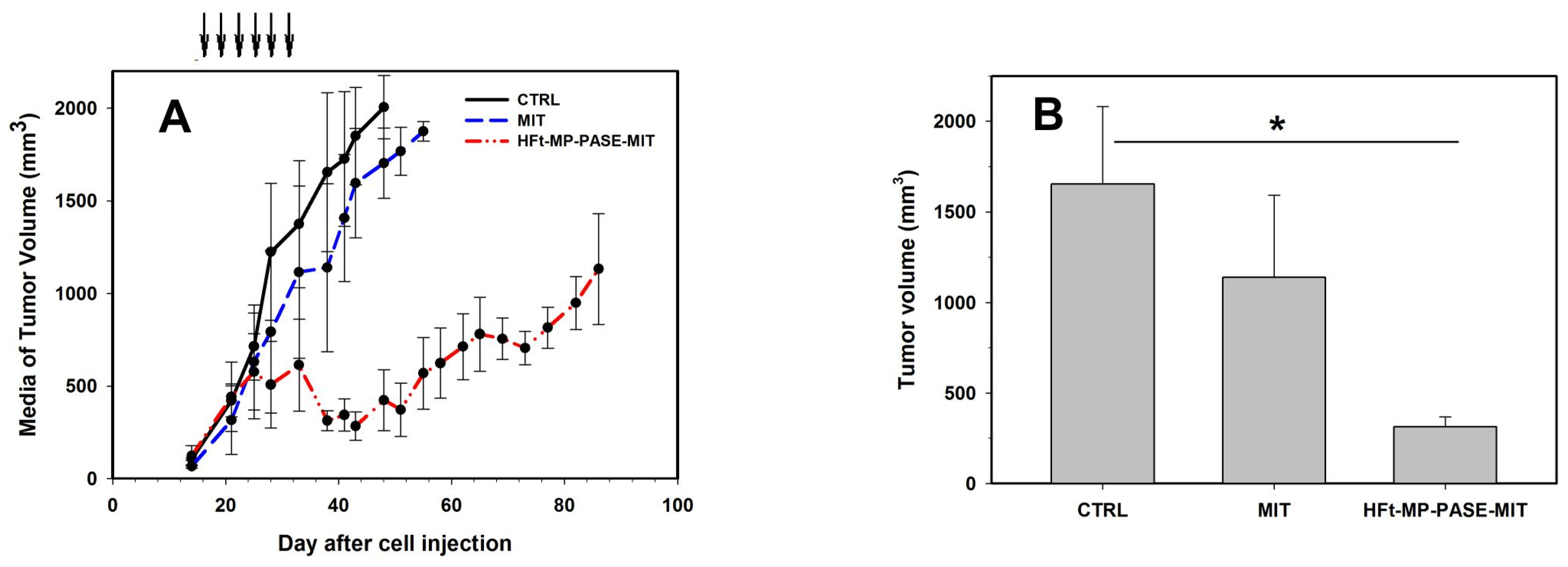
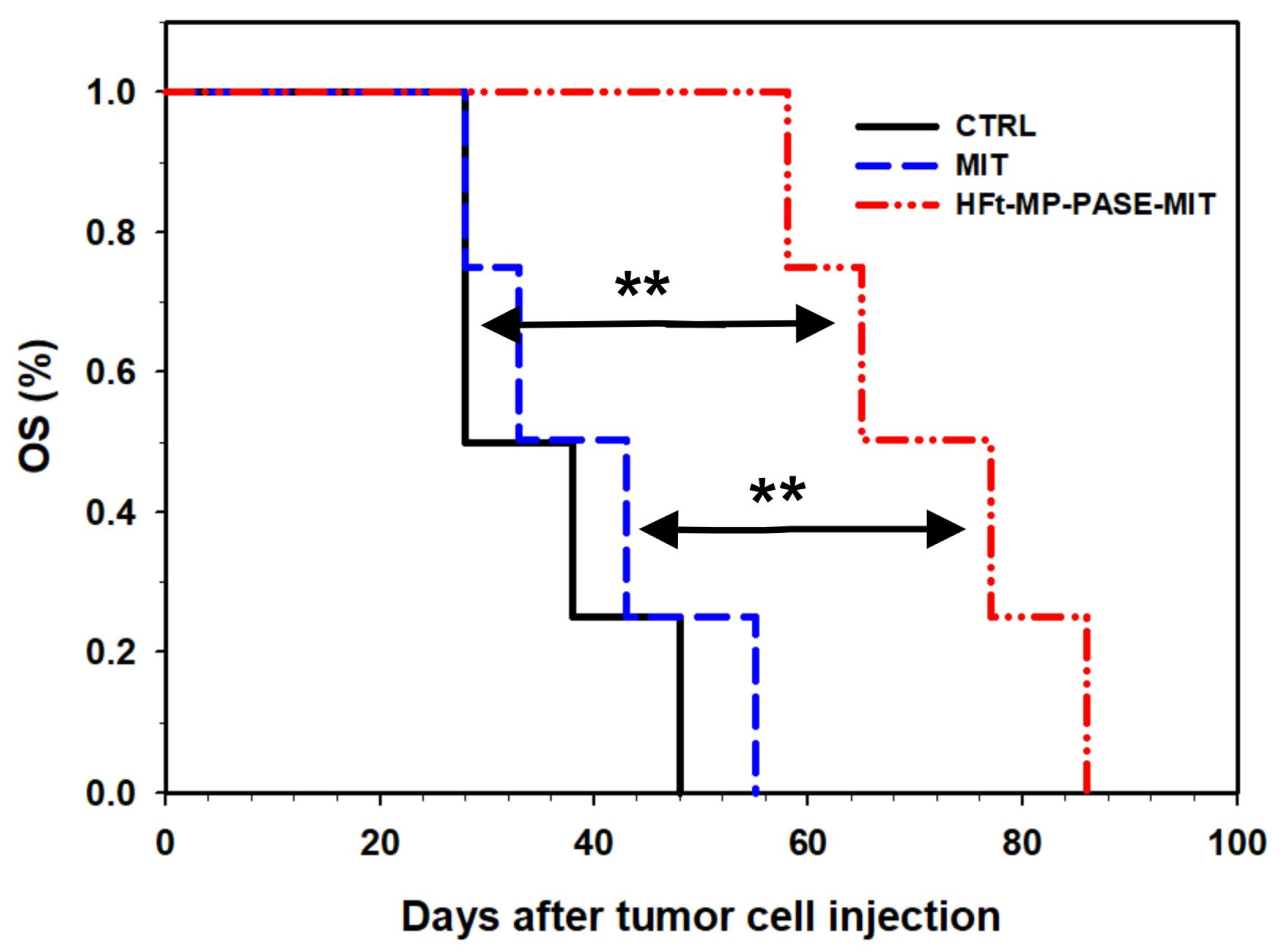
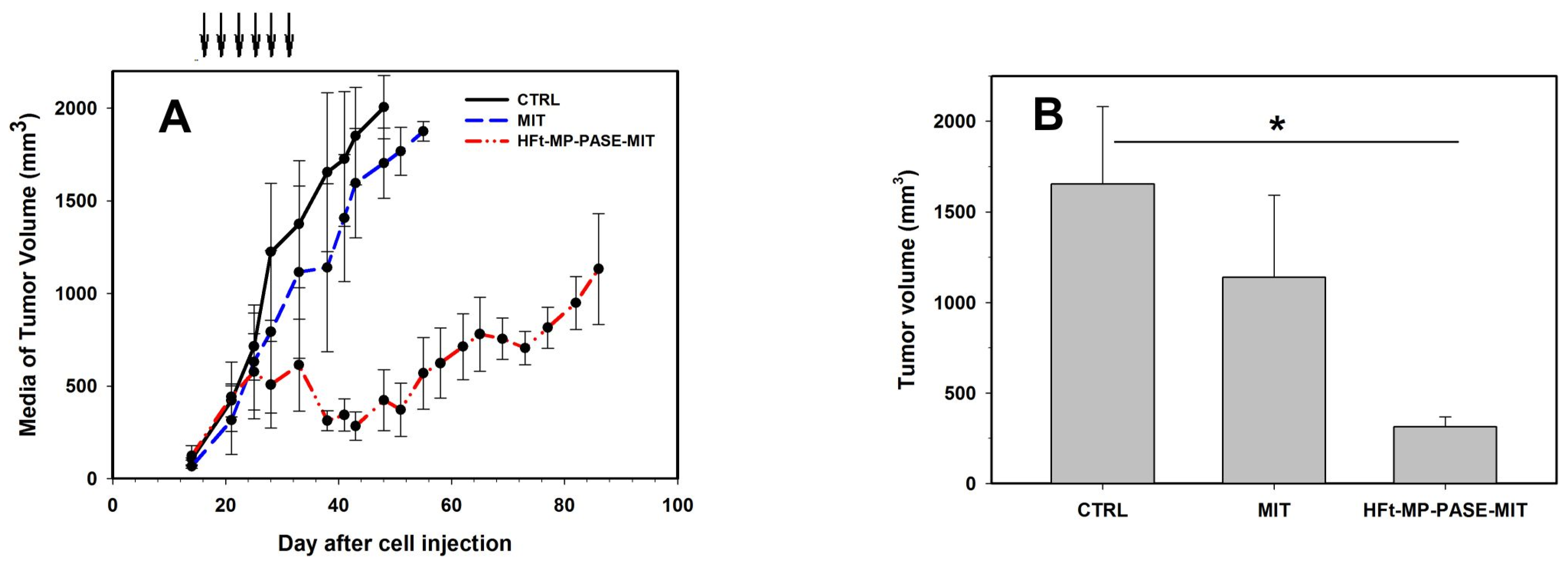
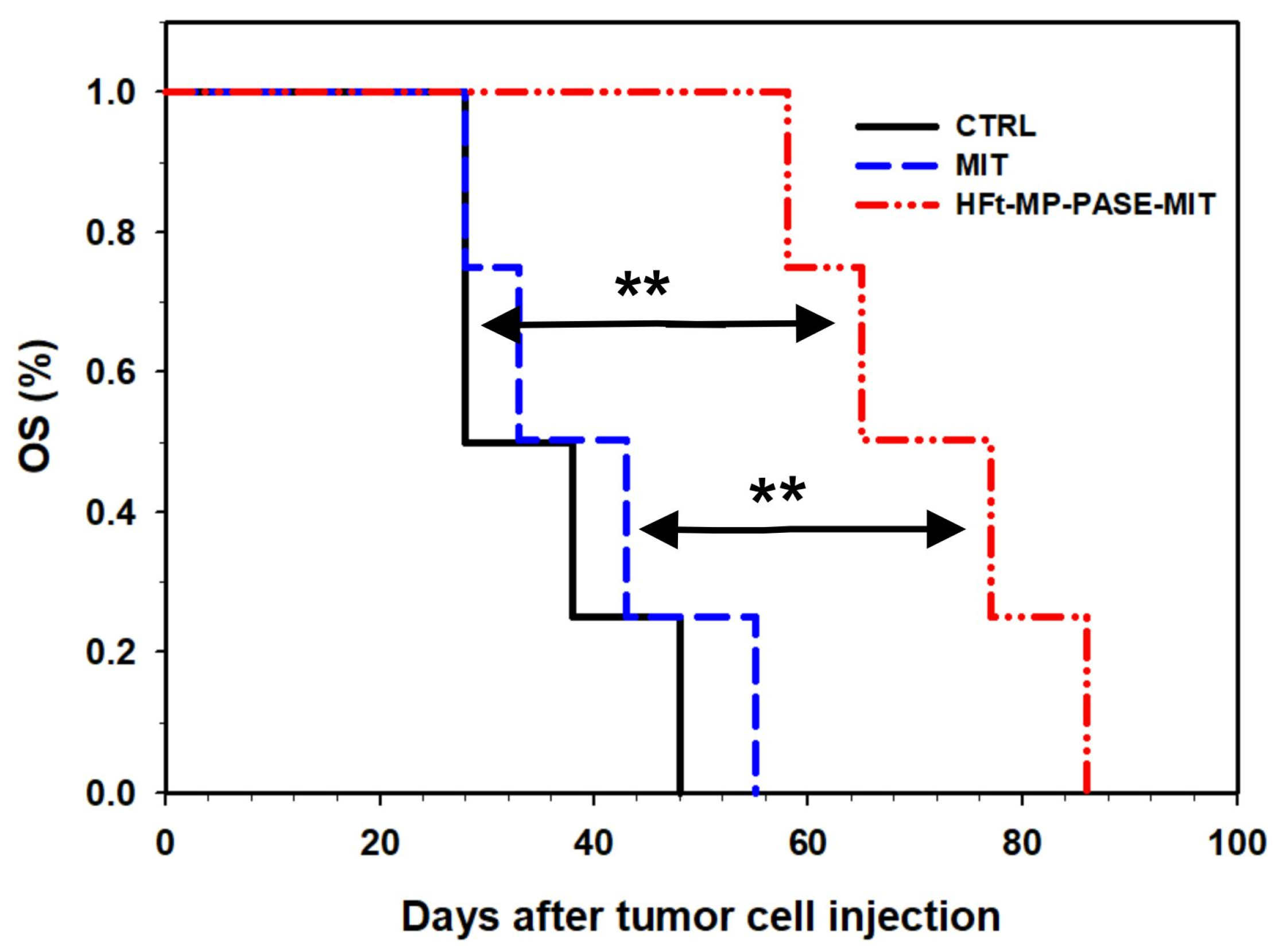
3.5. Therapeutic Efficacy In Vivo
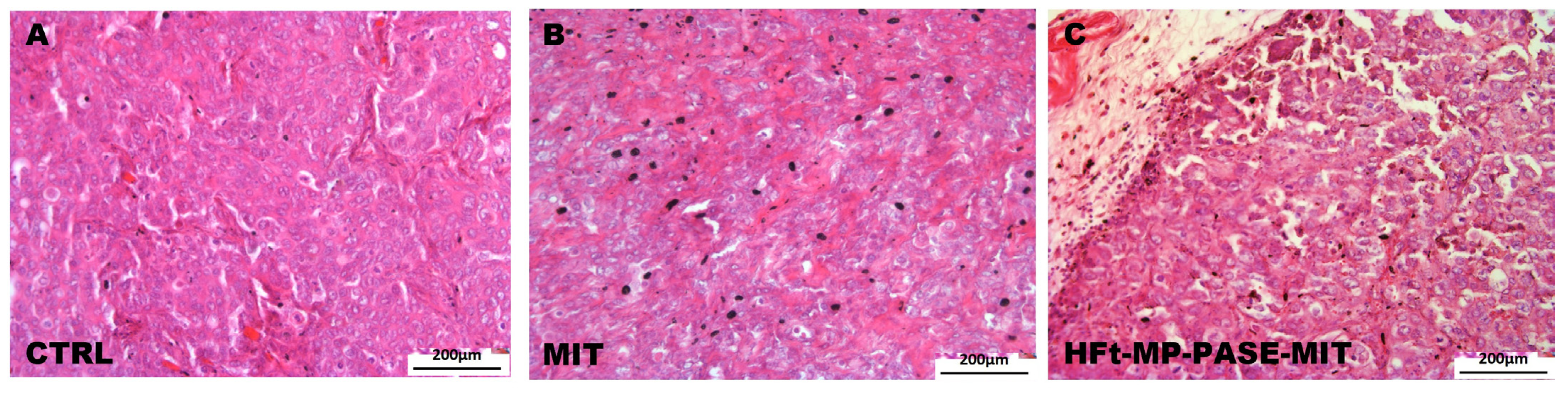
3.6. Histological Evaluation of Primary Tumor Mass
3.7. Histological Evaluation of Other Organs
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v56–v68. [Google Scholar] [CrossRef]
- Lv, J.; Li, P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 2019, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; Devroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1051. [Google Scholar] [CrossRef]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Fan, K.; Yan, X. Ferritin drug carrier (FDC) for tumor targeting therapy. J. Control. Release 2019, 311, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Falvo, E.; Tremante, E.; Arcovito, A.; Papi, M.; Elad, N.; Boffi, A.; Morea, V.; Conti, G.; Toffoli, G.; Fracasso, G.; et al. Improved Doxorubicin Encapsulation and Pharmacokinetics of Ferritin-Fusion Protein Nanocarriers Bearing Proline, Serine, and Alanine Elements. Biomacromolecules 2016, 17, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Falvo, E.; Arcovito, A.; Conti, G.; Cipolla, G.; Pitea, M.; Morea, V.; Damiani, V.; Sala, G.; Fracasso, G.; Ceci, P. Engineered human nanoferritin bearing the drug genz-644282 for cancer therapy. Pharmaceutics 2020, 12, 992. [Google Scholar] [CrossRef] [PubMed]
- Veroniaina, H.; Pan, X.; Wu, Z.; Qi, X. Apoferritin: A potential nanocarrier for cancer imaging and drug delivery. Expert Rev. Anticancer Ther. 2021, 21, 901–913. [Google Scholar] [CrossRef]
- Falvo, E.; Damiani, V.; Conti, G.; Boschi, F.; Messana, K.; Giacomini, P.; Milella, M.; De Laurenzi, V.; Morea, V.; Sala, G.; et al. High activity and low toxicity of a novel CD71-targeting nanotherapeutic named The-0504 on preclinical models of several human aggressive tumors. J. Exp. Clin. Cancer Res. 2021, 40, 63. [Google Scholar] [CrossRef]
- Falvo, E.; Malagrinò, F.; Arcovito, A.; Fazi, F.; Colotti, G.; Tremante, E.; Di Micco, P.; Braca, A.; Opri, R.; Giuffrè, A.; et al. The presence of glutamate residues on the PAS sequence of the stimuli-sensitive nano-ferritin improves in vivo biodistribution and mitoxantrone encapsulation homogeneity. J. Control. Release 2018, 275, 177–185. [Google Scholar] [CrossRef]
- Faulds, D.; Balfour, J.A.; Chrisp, P.; Langtry, H.D. Mitoxantrone. Drugs 1991, 41, 400–449. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Agency for Research on Cancer Some antiviral and antineoplastic drugs, and other pharmaceutical agents. IARC Monogr. Eval. Carcinog. Risks Hum. 2000, 76, 129–151. [Google Scholar]
- Riall, T.S.; Lillemoe, K.D. Underutilization of surgical resection in patients with localized pancreatic cancer. Ann. Surg. 2007, 246, 181–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal. Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Tesarova, B.; Musilek, K.; Rex, S.; Heger, Z. Taking advantage of cellular uptake of ferritin nanocages for targeted drug delivery. J. Control. Release 2020, 325, 176–190. [Google Scholar] [CrossRef]
- Habashy, H.O.; Powe, D.G.; Staka, C.M.; Rakha, E.A.; Ball, G.; Green, A.R.; Aleskandarany, M.; Paish, E.C.; Douglas MacMillan, R.; Nicholson, R.I.; et al. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res. Treat. 2010, 119, 283–293. [Google Scholar] [CrossRef]
- Adachi, M.; Kai, K.; Yamaji, K.; Ide, T.; Noshiro, H.; Kawaguchi, A.; Aishima, S. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology 2019, 75, 63–73. [Google Scholar] [CrossRef]
- Jamnongkan, W.; Thanan, R.; Techasen, A.; Namwat, N.; Loilome, W.; Intarawichian, P.; Titapun, A.; Yongvanit, P. Upregulation of transferrin receptor-1 induces cholangiocarcinoma progression via induction of labile iron pool. Tumor Biol. 2017, 39, 1010428317717655. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.J.; Attwood, K.; Sharma, N.J.; Gross, K.W.; Smith, G.J.; Xu, B.; Kauffman, E.C. Transferrin receptor 1 upregulation in primary tumor and downregulation in benign kidney is associated with progression and mortality in renal cell carcinoma patients. Oncotarget 2017, 8, 107052–107075. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Huszty, G.; Knaebel, H.P.; Hartel, M.; Büchler, M.W.; Schmidt, J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.C. The iron metabolism of neoplastic cells: Alterations that facilitate proliferation? Crit. Rev. Oncol. Hematol. 2002, 42, 65–78. [Google Scholar] [CrossRef]
- Steegmann-Olmedillas, J.L. The role of iron in tumour cell proliferation. Clin. Transl. Oncol. 2011, 13, 71–76. [Google Scholar] [CrossRef]
- Jeong, S.M.; Hwang, S.; Seong, R.H. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef]
- Burkhart, R.A.; Peng, Y.; Norris, Z.A.; Tholey, R.M.; Talbott, V.A.; Liang, Q.; Ai, Y.; Miller, K.; Lal, S.; Cozzitorto, J.A.; et al. Mitoxantrone Targets Human Ubiquitin-Specific Peptidase 11 (USP11) and Is a Potent Inhibitor of Pancreatic Cancer Cell Survival. Mol. Cancer Res. 2013, 11, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Hornung, A.; Poettler, M.; Friedrich, R.P.; Weigel, B.; Duerr, S.; Zaloga, J.; Cicha, I.; Alexiou, C.; Janko, C. Toxicity of Mitoxantrone-loaded Superparamagnetic Iron Oxide Nanoparticles in a HT-29 Tumour Spheroid Model. Anticancer Res. 2016, 36, 3093–3101. [Google Scholar]
- Yagublu, V.; Caliskan, N.; Lewis, A.L.; Jesenofsky, R.; Gasimova, L.; Löhr, J.M.; Keese, M. Treatment of experimental pancreatic cancer by doxorubicin-, mitoxantrone-, and irinotecan-drug eluting beads. Pancreatology 2012, 13, 79–87. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Wang, Y.; He, Y.; Feng, W.; Song, C. Pharmacokinetics, distribution and anti-tumor efficacy of liposomal mitoxantrone modified with a luteinizing hormone-releasing hormone receptor-specific peptide. Int. J. Nanomed. 2018, 13, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Tacchini, L.; Cairo, G. HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res. 1999, 27, 4223–4227. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Concentration | Early Apoptosis | Late Apoptosis | Viable Cells | Necrosis |
|---|---|---|---|---|
| GEM 15.2 mM | 10.01% ± 0.20 | 7.60% ± 0.50 | 58.00% ± 2.60 | 24.25% ± 2.25 |
| MIT 0.5 µM | 13.25% ± 1.85 | 6.35% ± 1.05 | 56.65% ± 1.45 | 23.80% ± 0.60 |
| HFt-MP-PASE-MIT 0.5 µM | 11.30% ± 1.00 | 4.95% ± 0.45 | 60.10% ± 4.90 | 23.70% ± 3.50 |
| 20 min | 40 min | 60 min | 90 min | 120 min | |
|---|---|---|---|---|---|
| HFt-MP-PASE-MIT vs. MIT | 133.07% ± 7.21 | 133.25% ± 7.37 | 139.49% ± 6.71 | 142.02% ± 7.00 | 138.37% ± 0.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, G.; Pitea, M.; Ossanna, R.; Opri, R.; Tisci, G.; Falvo, E.; Innamorati, G.; Ghanem, E.; Sbarbati, A.; Ceci, P.; et al. Mitoxantrone-Loaded Nanoferritin Slows Tumor Growth and Improves the Overall Survival Rate in a Subcutaneous Pancreatic Cancer Mouse Model. Biomedicines 2021, 9, 1622. https://doi.org/10.3390/biomedicines9111622
Conti G, Pitea M, Ossanna R, Opri R, Tisci G, Falvo E, Innamorati G, Ghanem E, Sbarbati A, Ceci P, et al. Mitoxantrone-Loaded Nanoferritin Slows Tumor Growth and Improves the Overall Survival Rate in a Subcutaneous Pancreatic Cancer Mouse Model. Biomedicines. 2021; 9(11):1622. https://doi.org/10.3390/biomedicines9111622
Chicago/Turabian StyleConti, Giamaica, Martina Pitea, Riccardo Ossanna, Roberta Opri, Giada Tisci, Elisabetta Falvo, Giulio Innamorati, Esther Ghanem, Andrea Sbarbati, Pierpaolo Ceci, and et al. 2021. "Mitoxantrone-Loaded Nanoferritin Slows Tumor Growth and Improves the Overall Survival Rate in a Subcutaneous Pancreatic Cancer Mouse Model" Biomedicines 9, no. 11: 1622. https://doi.org/10.3390/biomedicines9111622
APA StyleConti, G., Pitea, M., Ossanna, R., Opri, R., Tisci, G., Falvo, E., Innamorati, G., Ghanem, E., Sbarbati, A., Ceci, P., & Fracasso, G. (2021). Mitoxantrone-Loaded Nanoferritin Slows Tumor Growth and Improves the Overall Survival Rate in a Subcutaneous Pancreatic Cancer Mouse Model. Biomedicines, 9(11), 1622. https://doi.org/10.3390/biomedicines9111622