
Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies



Abstract
:1. Introduction
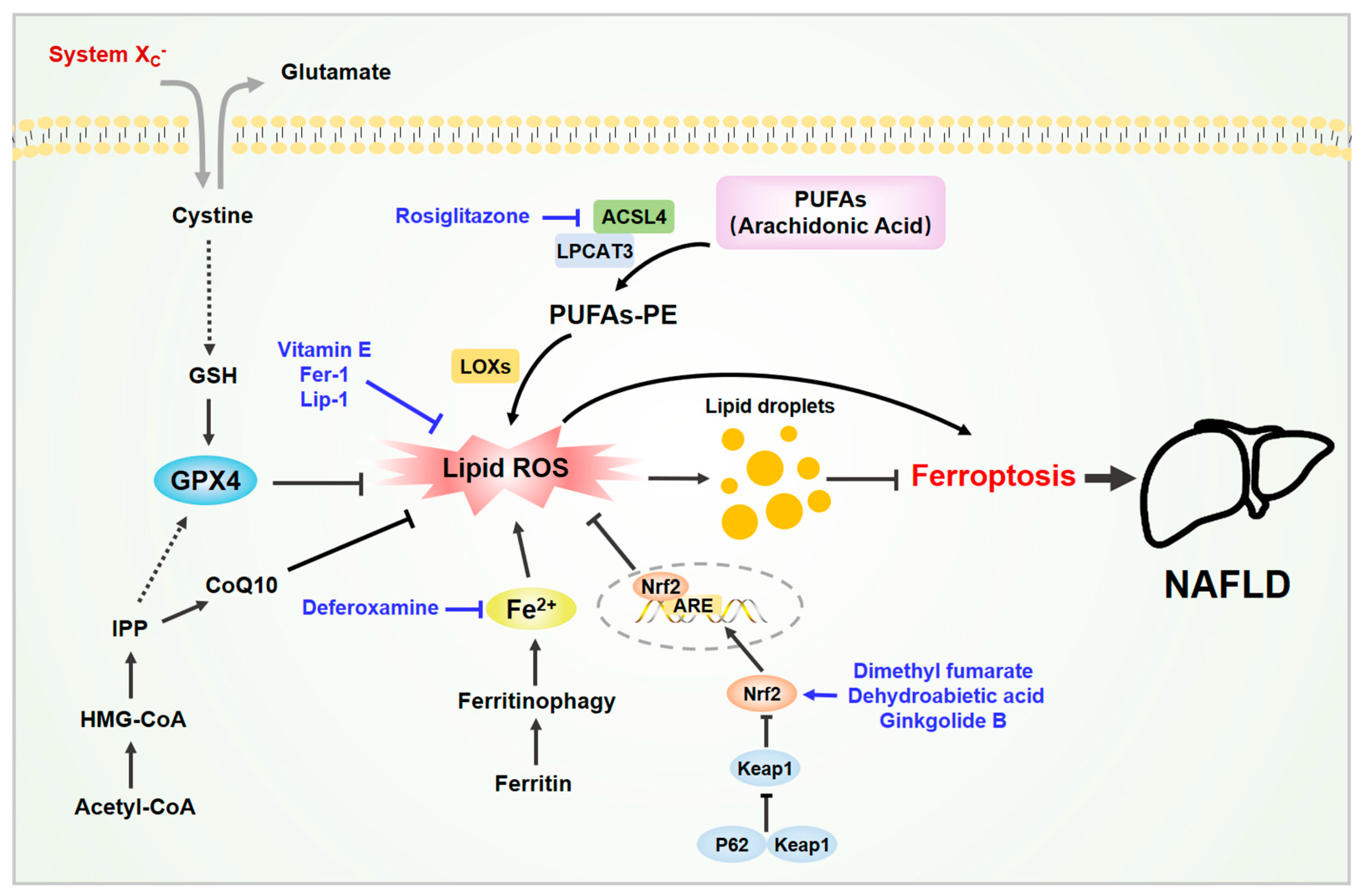
2. Ferroptosis and Its Mechanism
2.1. Iron Metabolism and Lipid Peroxidation
2.2. The System Xc−/GSH/GPX4 Axis
2.3. Other Related Signaling Metabolism
3. Ferroptosis and NAFLD
4. Mechanism of Ferroptosis in NAFLD
4.1. Decreased GPX4 Activity
4.2. Iron Overload
4.3. Lipid Peroxidation
4.4. ACSL4 Induction
4.5. Nrf2 Activation
4.6. Others
5. Drugs Targeting Ferroptosis in Liver Diseases
5.1. Ferroptosis Inducers
5.1.1. Erastin and Derivatives
5.1.2. Sulfasalazine
5.1.3. Sorafenib
5.1.4. Statins
5.1.5. Others
5.2. Ferroptosis Inhibitors
5.2.1. Fer-1
5.2.2. Vitamin E
5.2.3. Dimethyl Fumarate (DMF)
5.2.4. Rosiglitazone (ROSI)
5.2.5. Others
6. Conclusions and Future Directions
6.1. Detection of Lipid Peroxide Markers Related to Ferroptosis
6.2. The Role of Ferroptosis in Each Stage of NAFLD
6.3. Clinical Trials and Side Effects of Inhibiting Ferroptosis
6.4. The Role of Endotoxin in Regulating Ferroptosis in NAFLD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs. Nonalcoholic Steatohepatitis: A Systematic Review and Meta-analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Z.; Dai, Y.N.; Wang, Y.M.; Zhou, Q.Y.; Yu, C.H.; Li, Y.M. Prevalence of Nonalcoholic Fatty Liver Disease and Economy. Dig. Dis. Sci. 2015, 60, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnus, W.; Linkermann, A. The in vivo evidence for regulated necrosis. Immunol. Rev. 2017, 277, 128–149. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Buijs, M.; Doan, N.T.; van Rooden, S.; Versluis, M.J.; van Lew, B.; Milles, J.; van der Grond, J.; van Buchem, M.A. In vivo assessment of iron content of the cerebral cortex in healthy aging using 7-Tesla T2*-weighted phase imaging. Neurobiol. Aging 2017, 53, 20–26. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [Green Version]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Gascón, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.R.; Shah, Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar] [CrossRef] [Green Version]
- Salomao, M.A. Pathology of Hepatic Iron Overload. Clin. Liver Dis. 2021, 17, 232–237. [Google Scholar] [CrossRef]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br. J. Pharmacol. 2021, 178, 3783–3796. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stöckl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St. Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, D.M.; Anderson, G.J. The regulation of iron transport. BioFactors 2014, 40, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 508, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.A.; Anderson, E.J.; Gaudio, A.R.; et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch. Ophthalmol. 2004, 122, 1306–1314. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.J.; Pan, M.H.; Gong, H.B.; Lu, D.H.; Sun, J.; et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 17, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Canak, A.B.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System xc- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [Green Version]
- Kuganesan, N.; Dlamini, S.; McDaniel, J.; Tillekeratne, V.L.M.; Taylor, W.R. Identification and initial characterization of a potent inhibitor of ferroptosis. J. Cell. Biochem. 2021, 122, 413–424. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef]
- Reisman, S.A.; Yeager, R.L.; Yamamoto, M.; Klaassen, C.D. Increased Nrf2 activation in livers from keap1-knockdown mice Increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol. Sci. 2009, 108, 35–47. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Jawaid, Q.; Tortorelli, K.; LeClair, P.; Cobb, J.; Lambrecht, R.W.; Banner, B.F. Non-alcoholic steatohepatitis and iron: Increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 421–429. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Moscatiello, S.; Vanni, E.; Fracanzani, A.L.; Bugianesi, E.; Fargion, S.; Marchesini, G. Venesection for non-alcoholic fatty liver disease unresponsive to lifestyle counselling-a propensity score-adjusted observational study. QJM 2011, 104, 141–149. [Google Scholar] [CrossRef]
- Loguercio, C.; De Girolamo, V.; De Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Pia Di Marino, M.; et al. Non-alcoholic fatty liver disease in an area of southern italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, Y. Thymosin Beta 4 Is a Potential Regulator of Hepatic Stellate Cells. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2016; Volume 102, pp. 121–149. ISBN 9780128048184. [Google Scholar]
- Zhu, Z.; Zhang, Y.; Huang, X.; Can, L.; Zhao, X.; Wang, Y.; Xue, J.; Cheng, M.; Zhu, L. Thymosin beta 4 alleviates non-alcoholic fatty liver by inhibiting ferroptosis via up-regulation of GPX4. Eur. J. Pharmacol. 2021, 908, 174351. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, D.; Deng, C.; Wu, X.; Long, L.; Xiong, Y. Characterization of porcine ENO3: Genomic and cDNA structure, polymorphism and expression. Genet. Sel. Evol. 2008, 40, 563–579. [Google Scholar] [CrossRef]
- Lu, D.; Xia, Q.; Yang, Z.; Gao, S.; Sun, S.; Luo, X.; Li, Z.; Zhang, X.; Han, S.; Li, X.; et al. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via elevation of GPX4 expression and lipid accumulation. Ann. Transl. Med. 2021, 9, 661. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Wang, N.; Yao, X.; Jiang, L.; Jia, X.; Tao, Y.; Zhang, J.; Zhu, Y.; Yang, G.; et al. Ferroptosis mediated by the interaction between Mfn2 and IREα promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 2020, 188, 109824. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Xie, Z.; Li, E.-W.; Yuan, Y.; Fu, Y.; Wang, P.; Zhang, X.; Qiao, Y.; Xu, J.; Hölscher, C.; et al. Dehydroabietic acid improves nonalcoholic fatty liver disease through activating the Keap1/Nrf2-ARE signaling pathway to reduce ferroptosis. J. Nat. Med. 2021, 75, 540–552. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Gao, Q.; Shan, X.; Wang, J.; Lv, Z. Study on the attenuated effect of Ginkgolide B on ferroptosis in high fat diet induced nonalcoholic fatty liver disease. Toxicology 2020, 445, 152599. [Google Scholar] [CrossRef]
- Liu, B.; Yi, W.; Mao, X.; Yang, L.; Rao, C. Enoyl coenzyme A hydratase 1 alleviates nonalcoholic steatohepatitis in mice by suppressing hepatic ferroptosis. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E925–E937. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.H.; Cha, H.; Tang, J.; Lee, S.; Lee, S.H.; Le, B.; Redding, M.C.; Kim, S.; Batish, M.; Kong, B.C.; et al. The role of microRNA-33 as a key regulator in hepatic lipogenesis signaling and a potential serological biomarker for NAFLD with excessive dietary fructose consumption in C57BL/6N mice. Food Funct. 2021, 12, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623-633.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, M.; Li, Y.; Shen, M.; Kong, D.; Shao, J.; Ding, H.; Tan, S.; Chen, A.; Zhang, F.; et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 2020, 16, 1482–1505. [Google Scholar] [CrossRef]
- Chande, N.; Townsend, C.M.; Parker, C.E.; Macdonald, J.K. Azathioprine or 6-mercaptopurine for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2016, 2016, CD000545. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Estfan, B.; Byrne, M.; Kim, R. Sorafenib in Advanced Hepatocellular Carcinoma. Am. J. Clin. Oncol. 2013, 36, 319–324. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Mazière, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 2014. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The role of ferroptosis in cancer development and treatment response. Front. Pharmacol. 2018, 8, e02523. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, S.; Gu, H.; Lu, Q.; Jiang, W.; Pei, X.; Sun, Y.; Xu, H.; Wang, G.; Hao, K. The exposure-effect-toxicity correlation of docetaxel and magnesium isoglycyrrhizinate in non-small cell lung tumor-bearing mice. Biomed. Pharmacother. 2018, 97, 1000–1010. [Google Scholar] [CrossRef]
- Sui, M.; Jiang, X.; Chen, J.; Yang, H.; Zhu, Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2018, 106, 125–133. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wang, M.; Yu, H.M.; Han, F.X.; Wu, Q.S.; Cai, X.J.; Kurihara, H.; Chen, Y.X.; Li, Y.F.; He, R.R. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci. Biotechnol. Biochem. 2020, 84, 1621–1628. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y.; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, S.; Fu, Y.; Yan, L.; Feng, Y.; Chen, Y.; Wu, Y.; Deng, Y.; Zhang, G.; Chen, Z.; et al. Computational repositioning of dimethyl fumarate for treating alcoholic liver disease. Cell Death Dis. 2020, 11, 641. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lewin, T.M.; Coleman, R.A. Expression and Characterization of Recombinant Rat Acyl-CoA Synthetases 1, 4, and 5. J. Biol. Chem. 2001, 276, 24667–24673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Luo, Z.; Zhang, G.; Cao, D.; Li, D.; Ruan, H.; Ruan, B.H.; Su, L.; Xu, H. Click chemistry-based synthesis and anticancer activity evaluation of novel C-14 1,2,3-triazole dehydroabietic acid hybrids. Eur. J. Med. Chem. 2017, 138, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, Y.-G.; Lee, J.-Y.; Choi, D.-H.; Cho, Y.-U.; Shin, J.-M.; Park, J.S.; Lee, J.H.; Kim, W.G.; Seo, D.B.; et al. The natural phytochemical dehydroabietic acid is an anti-aging reagent that mediates the direct activation of SIRT1. Mol. Cell. Endocrinol. 2015, 412, 216–225. [Google Scholar] [CrossRef]
- Kim, E.; Kang, Y.G.; Kim, Y.J.; Lee, T.R.; Yoo, B.C.; Jo, M.; Kim, J.H.; Kim, J.H.; Kim, D.; Cho, J.Y. Dehydroabietic acid suppresses inflammatory response via suppression of Src-, Syk-, and TAK1-mediated pathways. Int. J. Mol. Sci. 2019, 20, 1593. [Google Scholar] [CrossRef] [Green Version]
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [Green Version]
- Mostafa Abd El-Aal, H.A.H. Lipid Peroxidation End-Products as a Key of Oxidative Stress: Effect of Antioxidant on Their Production and Transfer of Free Radicals. In Lipid Peroxidation; IntechOpen: London, UK, 2012. [Google Scholar]
- Horoz, M.; Bolukbas, C.; Bolukbas, F.F.; Sabuncu, T.; Aslan, M.; Sarifakiogullari, S.; Gunaydin, N.; Erel, O. Measurement of the total antioxidant response using a novel automated method in subjects with nonalcoholic steatohepatitis. BMC Gastroenterol. 2005, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, Z.; Gibson, D.L.; Hekmatdoost, A. Nonalcoholic fatty liver disease, the gut microbiome, and diet. Adv. Nutr. 2017, 8, 240–252. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugino, K.; Dohi, K.; Yamada, K.; Kawasaki, T. The role of lipid peroxidation in endotoxin-induced hepatic damage and the protective effect of antioxidants. Surgery 1987, 101, 746–752. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drugs | Ferroptosis | Target | Mechanism |
|---|---|---|---|
| Erastin [43,44] | Inducer | System Xc− | Inhibits system Xc−, resulting in GSH depletion |
| Sulfasalazine [47,48,49] | Inducer | System Xc− | Inhibits system Xc−, resulting in GSH depletion |
| Sorafenib [50,51,52] | Inducer | System Xc− | Inhibits system Xc−, resulting in GSH depletion |
| Statins [55] | Inducer | HMG-CoA reductase | Depletes CoQ10, resulting in lipid peroxidation |
| Artesunate [56,57] | Inducer | Ferritinophagy | Activates ferritinophagy |
| Magnesium isoglycyrrhizinate [59] | Inducer | HO-1 | Up-regulates HO-1 |
| Fer-1 [5,11,39,60] | Inhibitor | PUFAs | Inhibits lipid peroxidation |
| Vitamin E [61,62,63] | Inhibitor | PUFAs | Inhibits lipid peroxidation |
| Dimethyl fumarate [64] | Inhibitor | Nrf2 | Activates Nrf2, leading to reducing lipid peroxidation |
| Rosiglitazone [19,65] | Inhibitor | ACSL4 | Inhibits ACSL4, leading to reducing lipid peroxidation |
| Dehydroabietic acid [69] | Inhibitor | Nrf2 | Activates Nrf2, leading to reducing lipid peroxidation |
| Ginkgolide B [70] | Inhibitor | Nrf2 | Activates Nrf2, leading to reducing lipid peroxidation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhang, E.; Hu, H. Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies. Biomedicines 2021, 9, 1660. https://doi.org/10.3390/biomedicines9111660
Zhang H, Zhang E, Hu H. Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies. Biomedicines. 2021; 9(11):1660. https://doi.org/10.3390/biomedicines9111660
Chicago/Turabian StyleZhang, Han, Enxiang Zhang, and Hongbo Hu. 2021. "Role of Ferroptosis in Non-Alcoholic Fatty Liver Disease and Its Implications for Therapeutic Strategies" Biomedicines 9, no. 11: 1660. https://doi.org/10.3390/biomedicines9111660