
B Lymphocyte-Deficiency in Mice Causes Vascular Dysfunction by Inducing Neutrophilia

 , , ,
, , ,  ,
,  , and
, and 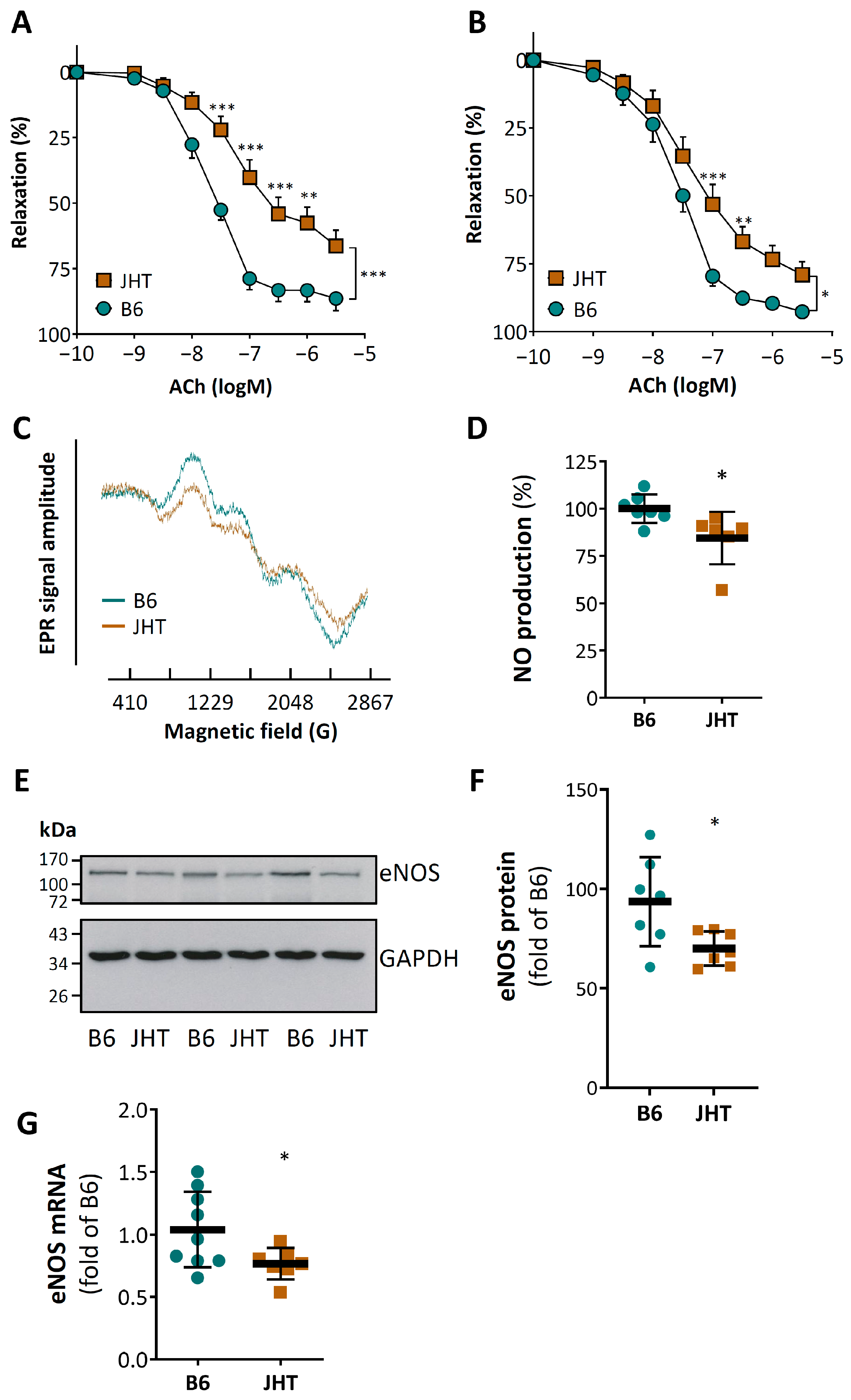{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Neutrophil Depletion
2.3. B Cell Transfer Experiments
2.4. Myograph Experiments
2.5. Electron Paramagnetic Resonance (EPR)
2.6. Measurement of ROS Production
2.7. Gene Expression Analyses
2.8. Western Blot Analyses
2.9. Flow Cytometric Analyses
2.10. Measurement of Neopterin in Mouse Serum
2.11. Statistical Analysis
3. Results
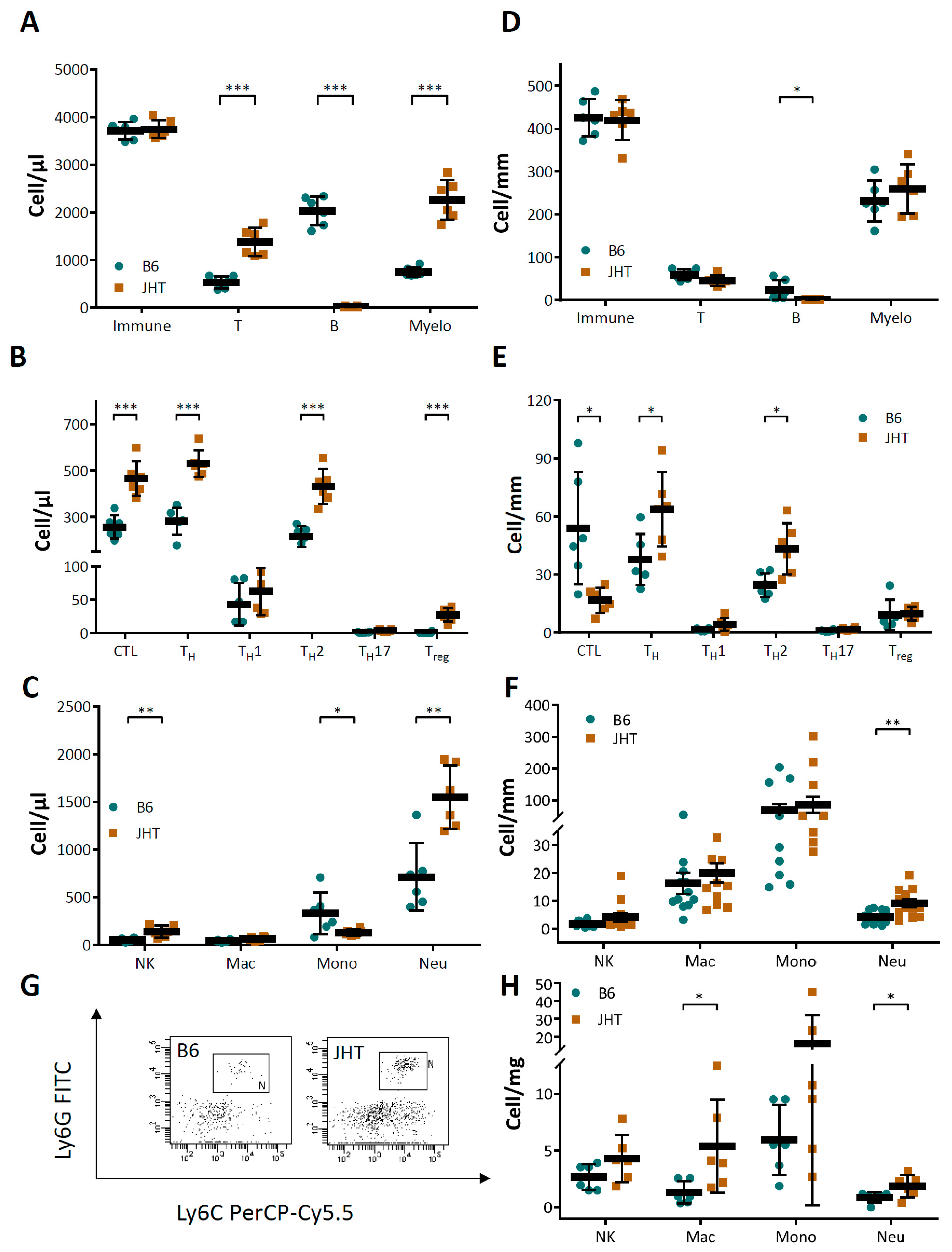
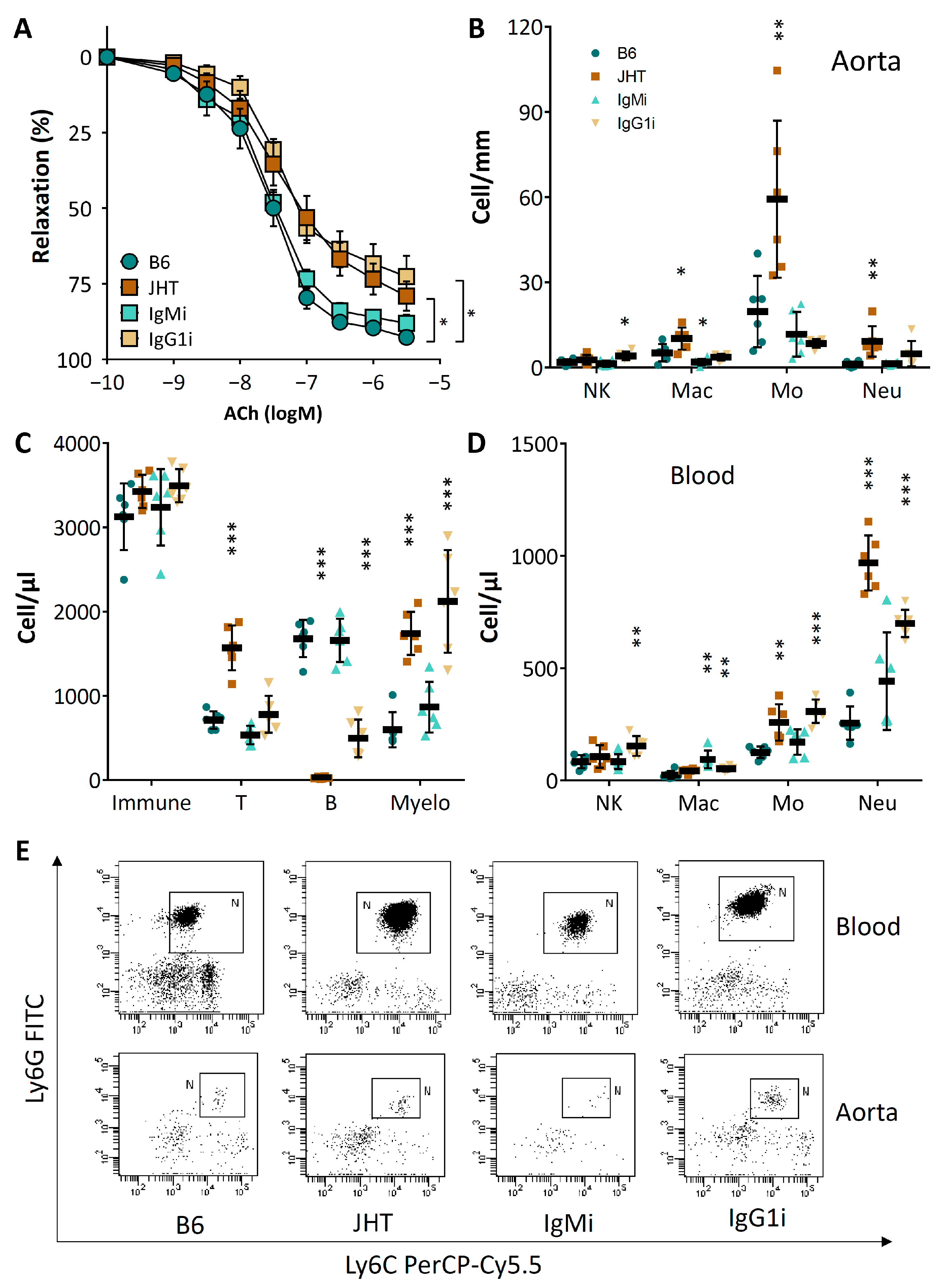
3.1. B Cell-Deficiency in Mice Leads to Vascular Dysfunction and Reduced NO Production
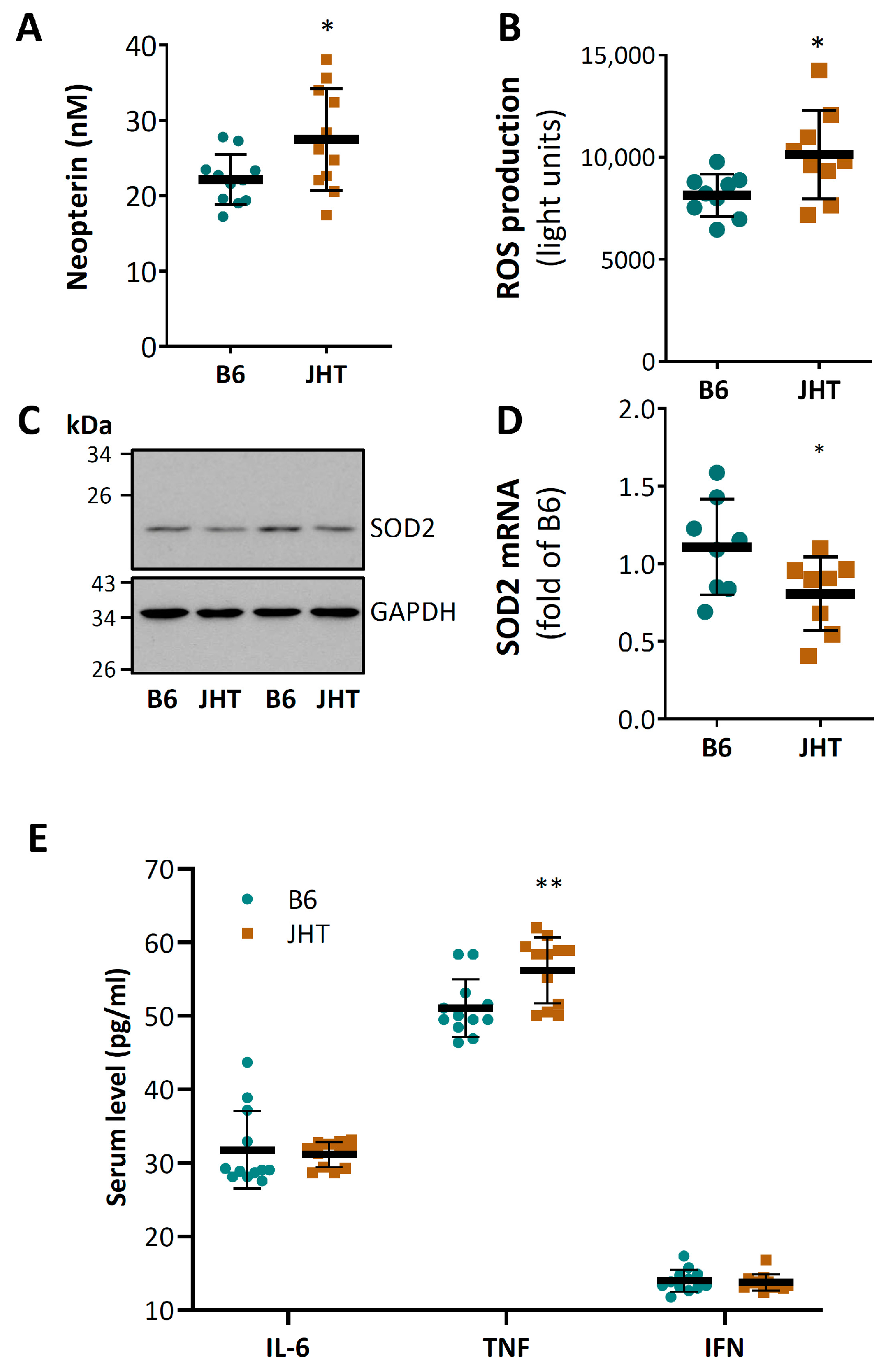
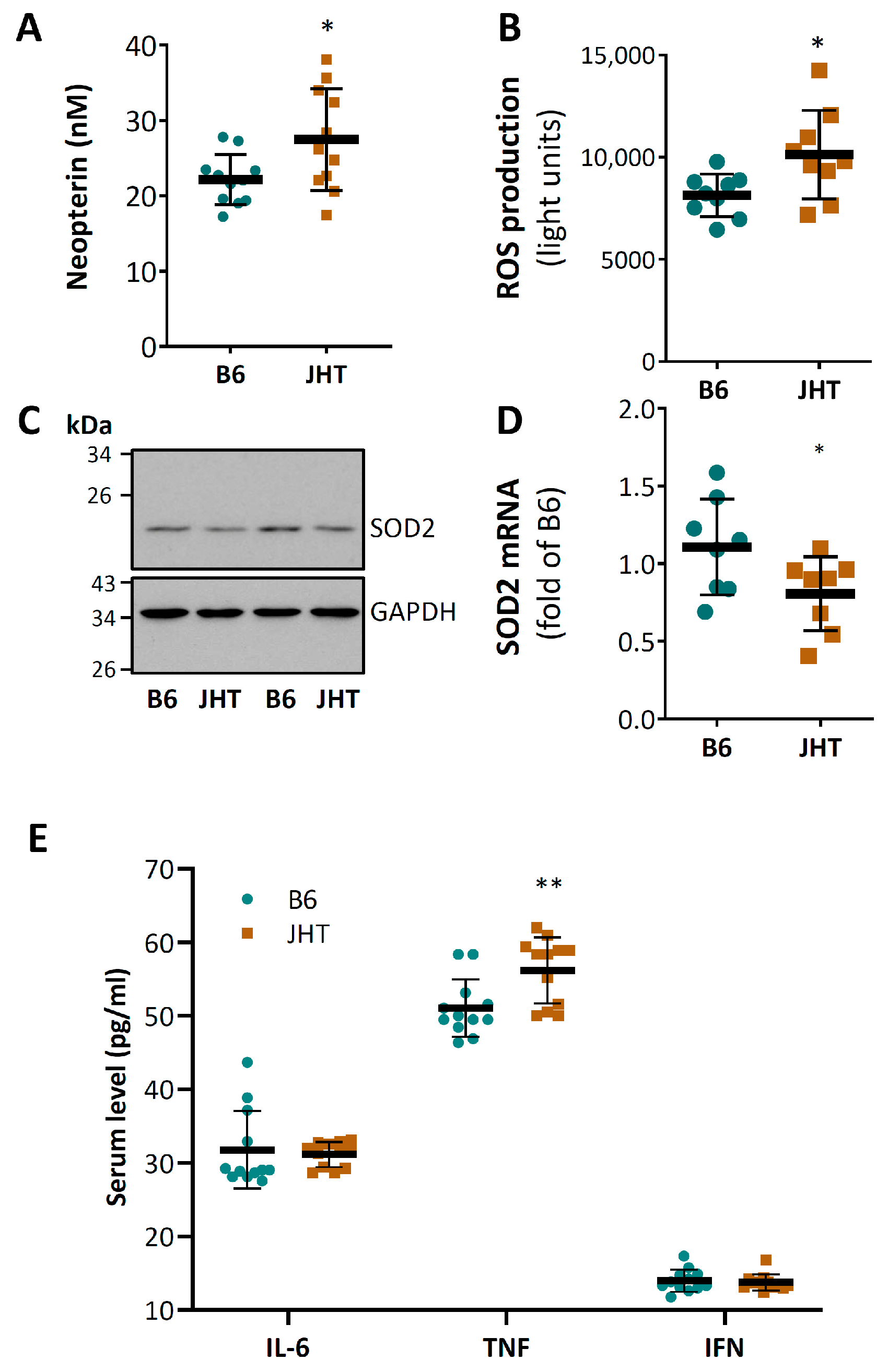
3.2. B Cell-Deficiency in Mice Leads to Vascular Oxidative Stress and Inflammation
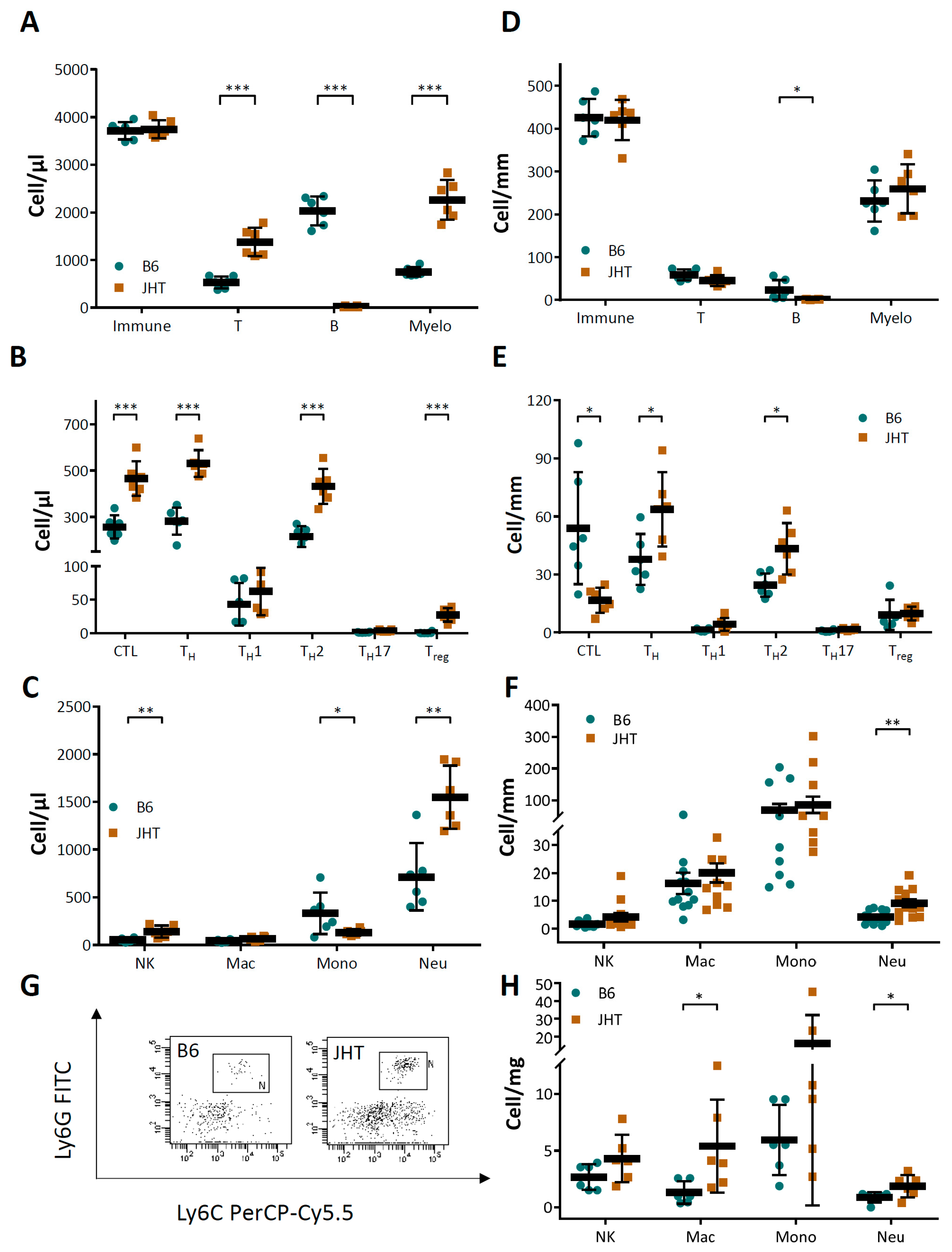
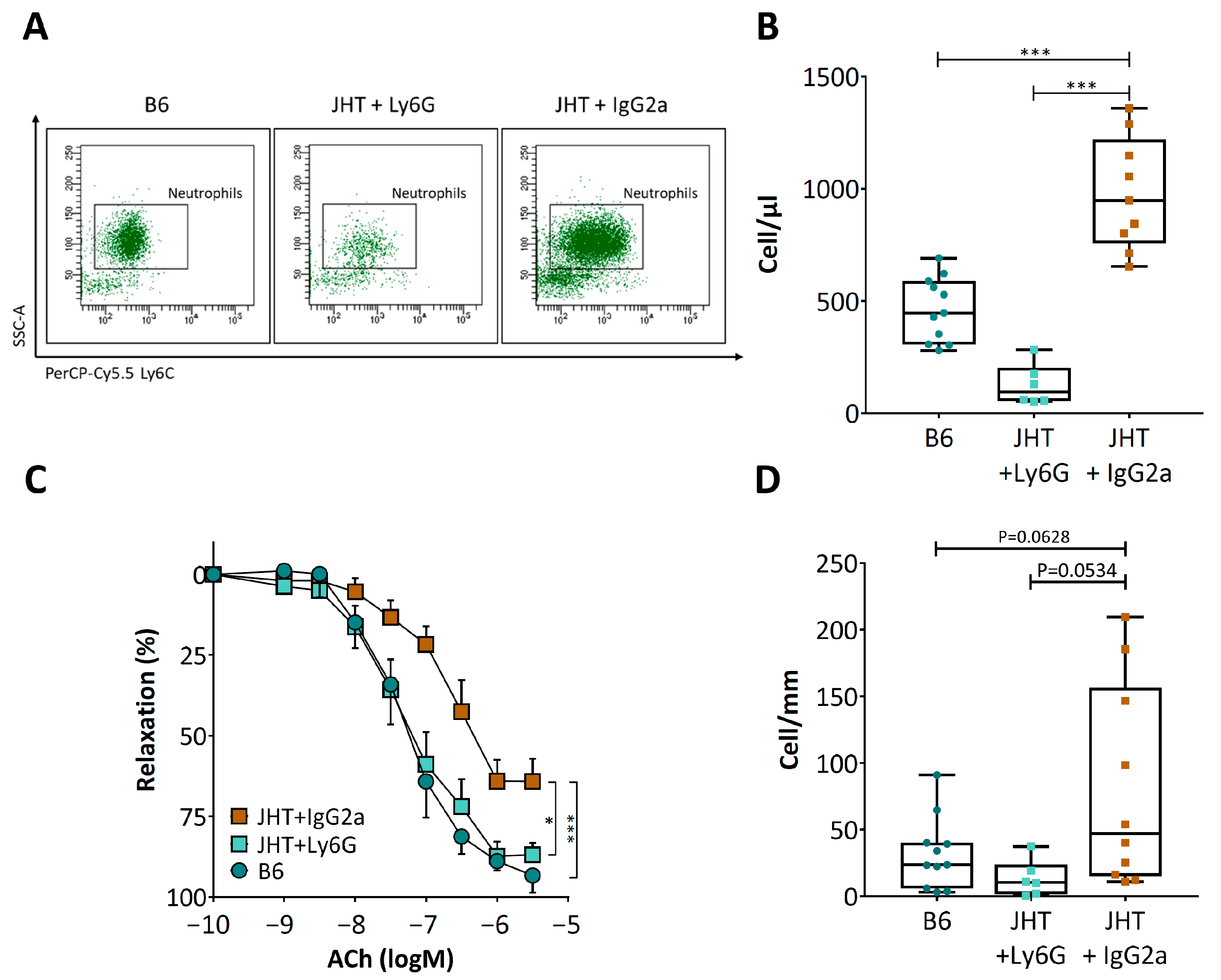
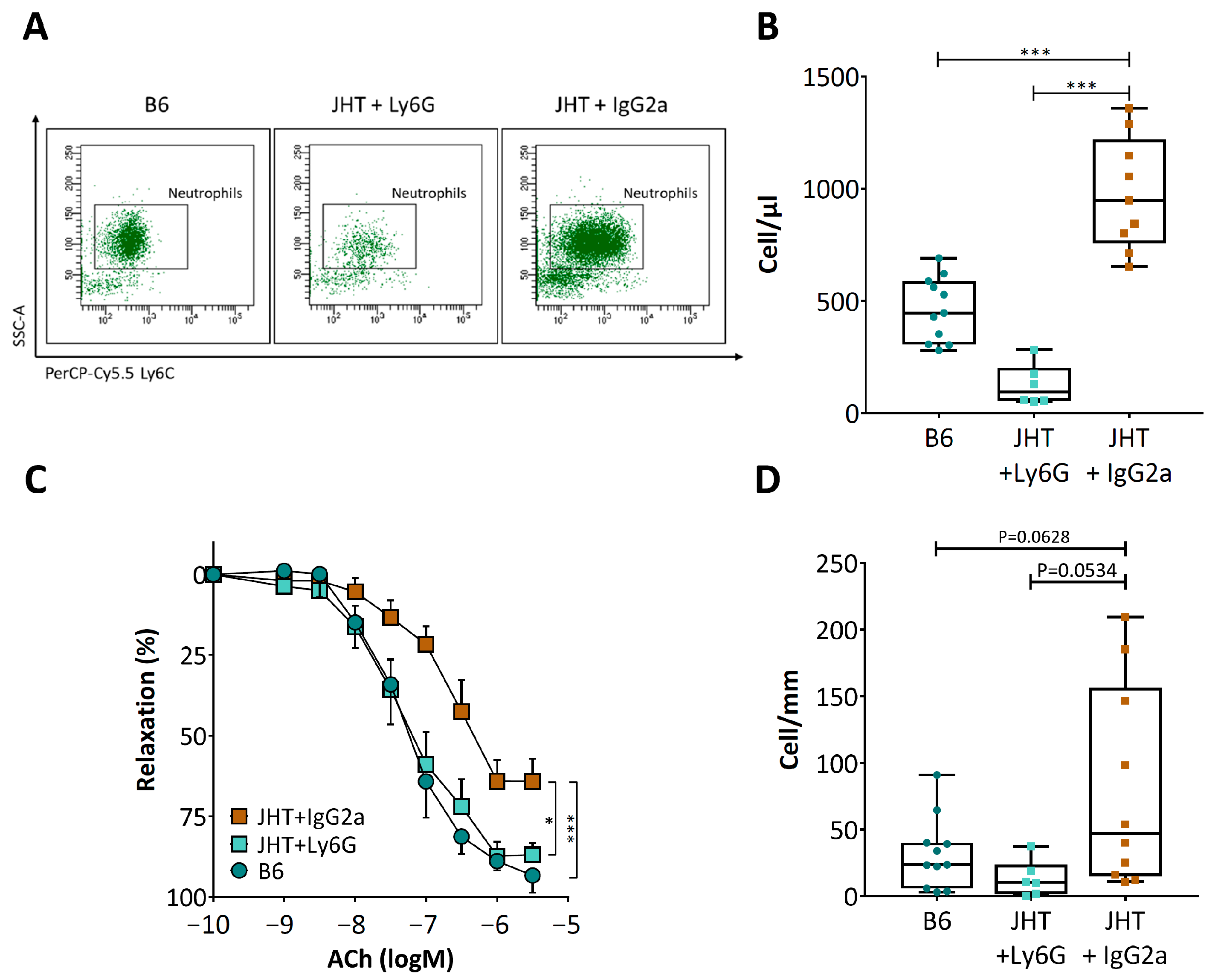
3.3. Vascular Dysfunction in B Cell-Deficiency in Mice Is Correlated with Neutrophilia
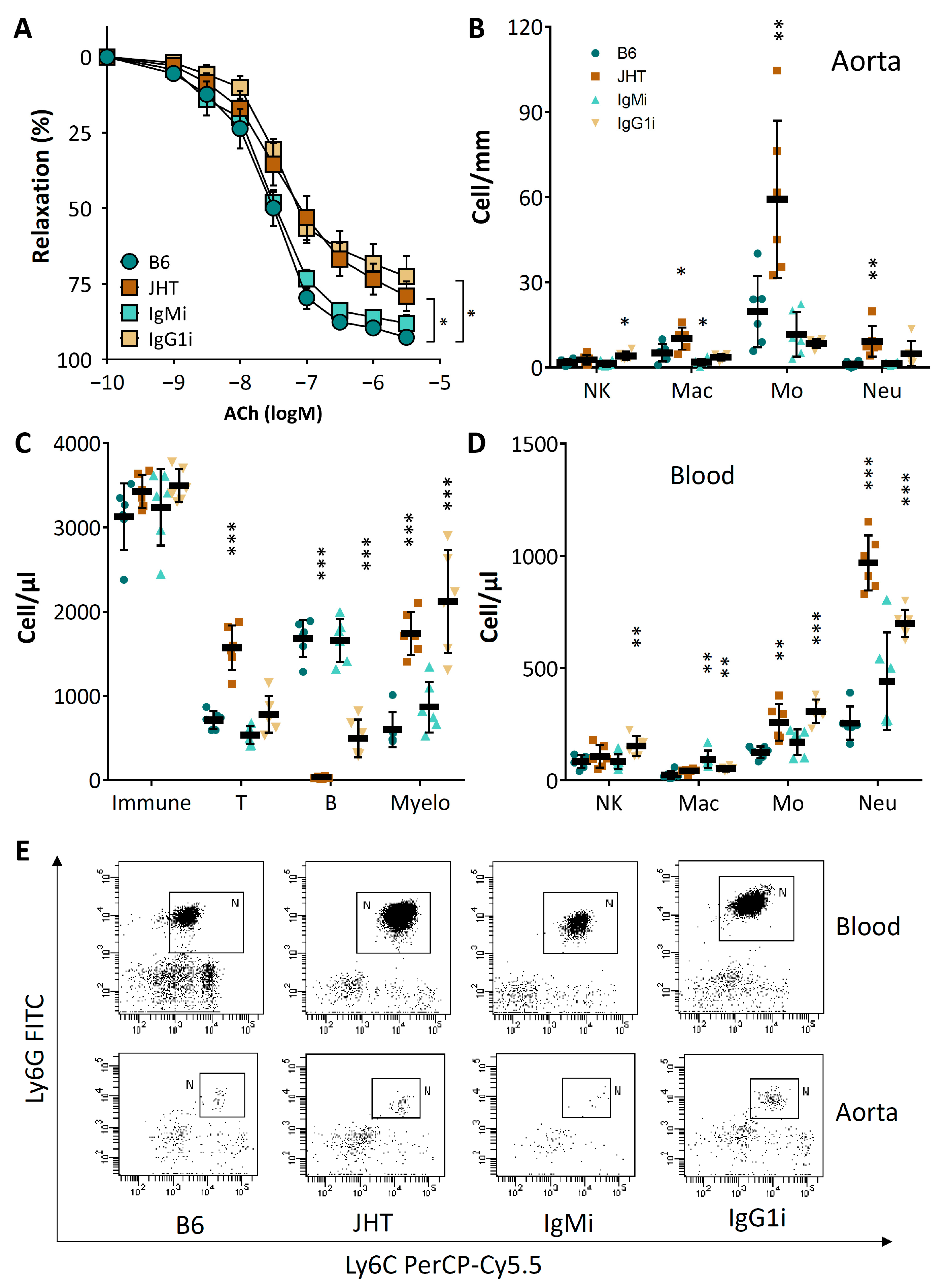
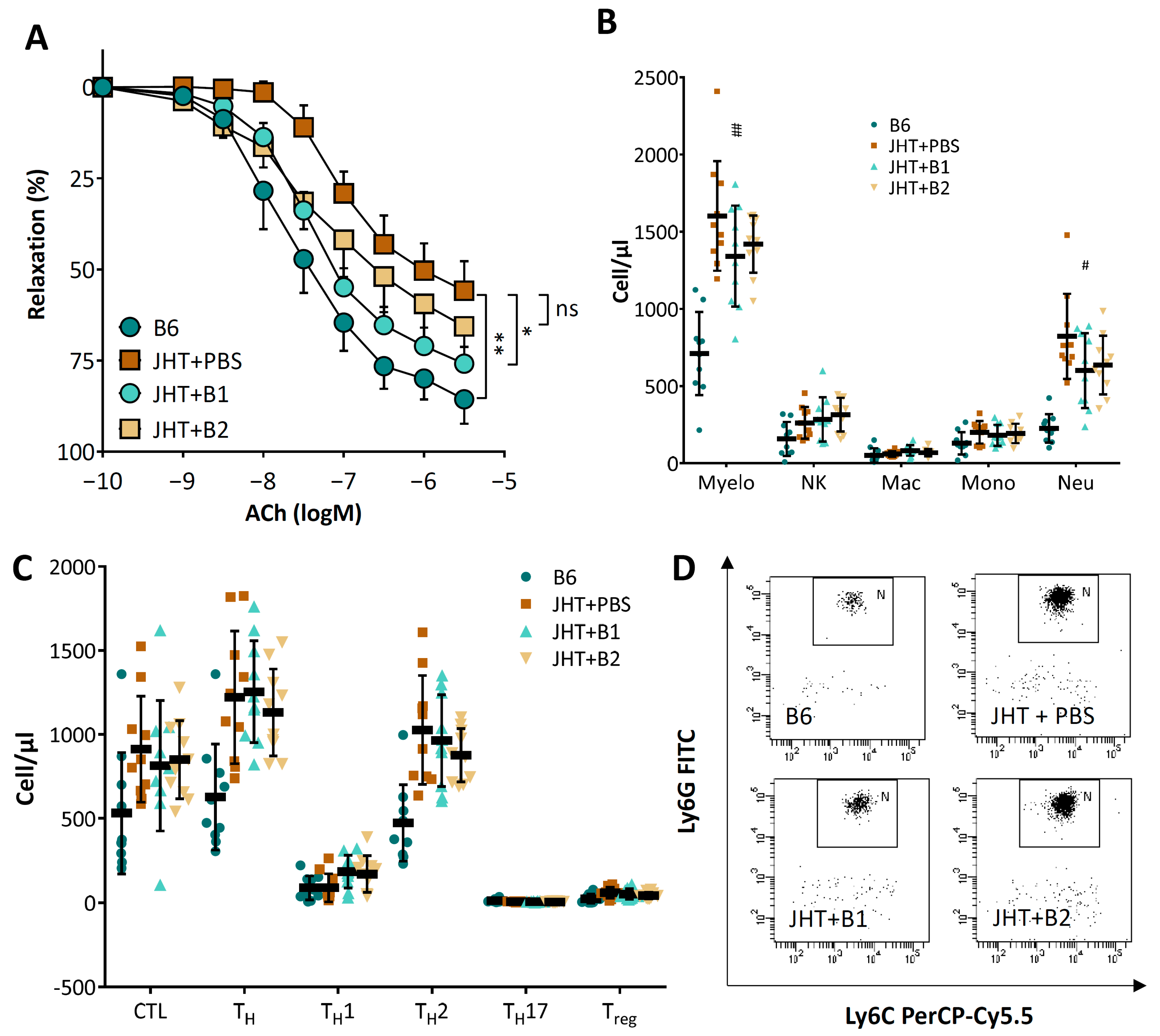
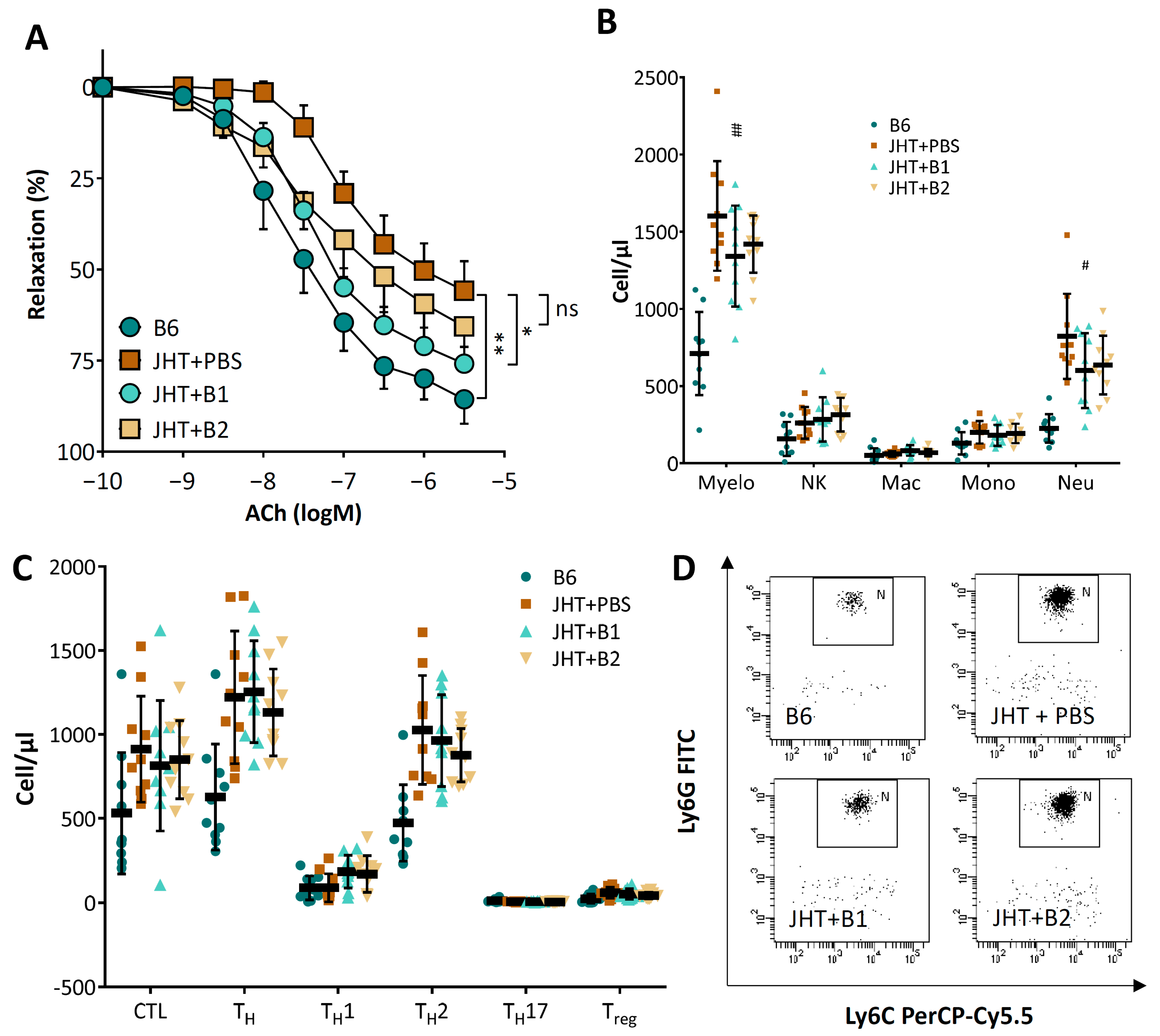
3.4. Vascular Dysfunction in B Cell-Deficient Mice Is Improved by B-1 Cell Transfer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Availability of Data and Material
References
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Porsch, F.; Binder, C.J. Impact of B-Cell-Targeted Therapies on Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M.; Bender, T.P.; McNamara, C.A. B cell subsets in atherosclerosis. Front. Immunol. 2012, 3, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkina, E.; Kadl, A.; Sanders, J.; Varughese, D.; Sarembock, I.J.; Ley, K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 2006, 203, 1273–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamze, M.; Desmetz, C.; Berthe, M.L.; Roger, P.; Boulle, N.; Brancherau, P.; Picard, E.; Guzman, C.; Tolza, C.; Guglielmi, P. Characterization of resident B cells of vascular walls in human atherosclerotic patients. J. Immunol. 2013, 191, 3006–3016. [Google Scholar] [CrossRef] [Green Version]
- Farias-Itao, D.S.; Pasqualucci, C.A.; Nishizawa, A.; da Silva, L.F.F.; Campos, F.M.; Bittencourt, M.S.; da Silva, K.C.S.; Leite, R.E.P.; Grinberg, L.T.; Ferretti-Rebustini, R.E.L.; et al. B Lymphocytes and Macrophages in the Perivascular Adipose Tissue Are Associated With Coronary Atherosclerosis: An Autopsy Study. J. Am. Heart Assoc. 2019, 8, e013793. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Krishnamurthi, S.; Kanellakis, P.; Agrotis, A.; Tipping, P.; Bobik, A.; Toh, B.H. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ. Res. 2011, 109, 830–840. [Google Scholar] [CrossRef]
- Rosenfeld, S.M.; Perry, H.M.; Gonen, A.; Prohaska, T.A.; Srikakulapu, P.; Grewal, S.; Das, D.; McSkimming, C.; Taylor, A.M.; Tsimikas, S.; et al. B-1b Cells Secrete Atheroprotective IgM and Attenuate Atherosclerosis. Circ. Res. 2015, 117, e28–e39. [Google Scholar] [CrossRef] [Green Version]
- Upadhye, A.; Srikakulapu, P.; Gonen, A.; Hendrikx, S.; Perry, H.M.; Nguyen, A.; McSkimming, C.; Marshall, M.A.; Garmey, J.C.; Taylor, A.M.; et al. Diversification and CXCR4-Dependent Establishment of the Bone Marrow B-1a Cell Pool Governs Atheroprotective IgM Production Linked to Human Coronary Atherosclerosis. Circ. Res. 2019, 125, e55–e70. [Google Scholar] [CrossRef]
- Doring, Y.; Jansen, Y.; Cimen, I.; Aslani, M.; Gencer, S.; Peters, L.J.F.; Duchene, J.; Weber, C.; van der Vorst, E.P.C. B-Cell-Specific CXCR4 Protects Against Atherosclerosis Development and Increases Plasma IgM Levels. Circ. Res. 2020, 126, 787–788. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Theurl, I.; Gerhardt, L.M.; Robbins, C.S.; Weber, G.F.; Gonen, A.; Iwamoto, Y.; Degousee, N.; Holderried, T.A.; Winter, C.; et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation 2014, 129, 1677–1687. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Sage, A.P.; Mallat, Z.; Tedgui, A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ. Res. 2014, 114, 1640–1660. [Google Scholar] [CrossRef]
- Morris-Rosenfeld, S.; Lipinski, M.J.; McNamara, C.A. Understanding the role of B cells in atherosclerosis: Potential clinical implications. Expert Rev. Clin. Immunol. 2014, 10, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, A.P.; Nus, M.; Baker, L.L.; Finigan, A.J.; Masters, L.M.; Mallat, Z. Regulatory B Cell-Specific Interleukin-10 Is Dispensable for Atherosclerosis Development in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1770–1773. [Google Scholar] [CrossRef] [Green Version]
- Strom, A.C.; Cross, A.J.; Cole, J.E.; Blair, P.A.; Leib, C.; Goddard, M.E.; Rosser, E.C.; Park, I.; Nilsson, A.H.; Nilsson, J.; et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb. Haemost. 2015, 114, 835–847. [Google Scholar]
- Douna, H.; Amersfoort, J.; Schaftenaar, F.H.; Kroon, S.; van Puijvelde, G.H.M.; Kuiper, J.; Foks, A.C. Bidirectional effects of IL-10(+) regulatory B cells in Ldlr(-/-) mice. Atherosclerosis 2019, 280, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Paradis, P.; Schiffrin, E.L. Role of immune cells in hypertension. Br. J. Pharmacol. 2019, 176, 1818–1828. [Google Scholar] [CrossRef]
- Norlander, A.E.; Madhur, M.S.; Harrison, D.G. The immunology of hypertension. J. Exp. Med. 2018, 215, 21–33. [Google Scholar] [CrossRef]
- Mikolajczyk, T.P.; Guzik, T.J. Adaptive Immunity in Hypertension. Curr. Hypertens. Rep. 2019, 21, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.; Sobey, C.G.; Lieu, M.; Ferens, D.; Kett, M.M.; Diep, H.; Kim, H.A.; Krishnan, S.M.; Lewis, C.V.; Salimova, E.; et al. Role for B Cells in the Development of Angiotensin II-Dependent Hypertension. Hypertension 2015, 66, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Barati, M.T.; Powell, D.W.; Turbeville, H.R.; Ryan, M.J. Plasma Cell Depletion Attenuates Hypertension in an Experimental Model of Autoimmune Disease. Hypertension 2018, 71, 719–728. [Google Scholar] [CrossRef]
- Gu, H.; Zou, Y.R.; Rajewsky, K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 1993, 73, 1155–1164. [Google Scholar] [CrossRef]
- Waisman, A.; Kraus, M.; Seagal, J.; Ghosh, S.; Melamed, D.; Song, J.; Sasaki, Y.; Classen, S.; Lutz, C.; Brombacher, F.; et al. IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Ig alpha/beta. J. Exp. Med. 2007, 204, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Croxford, A.L.; Demircik, F. New tools to study the role of B cells in cytomegalovirus infections. Med. Microbiol. Immunol. 2008, 197, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.J.; Andrews, N.; Ball, D.; Bellantuono, I.; Gray, J.; Hachoumi, L.; Holmes, A.; Latcham, J.; Petrie, A.; Potter, P.; et al. Does age matter? The impact of rodent age on study outcomes. Lab. Anim. 2017, 51, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Dittel, B.N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 2010, 2010, 488. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Forstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Munzel, T.; Daiber, A.; Forstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Witte, K.; August, M.; Brausch, I.; Godtel-Armbrust, U.; Habermeier, A.; Closs, E.I.; Oelze, M.; Munzel, T.; Forstermann, U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006, 47, 2536–2544. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; August, M.; Baldus, S.; Wendt, M.; Oelze, M.; Sydow, K.; Kleschyov, A.L.; Munzel, T. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic. Biol. Med. 2004, 36, 101–111. [Google Scholar] [CrossRef]
- Kossmann, S.; Lagrange, J.; Jackel, S.; Jurk, K.; Ehlken, M.; Schonfelder, T.; Weihert, Y.; Knorr, M.; Brandt, M.; Xia, N.; et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci. Transl. Med. 2017, 9, eaah4923. [Google Scholar] [CrossRef] [PubMed]
- Flavall, E.A.; Crone, E.M.; Moore, G.A.; Gieseg, S.P. Dissociation of neopterin and 7,8-dihydroneopterin from plasma components before HPLC analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 863, 167–171. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Sage, A.P.; Mallat, Z.; Binder, C.J. Targeting B cells in atherosclerosis: Closing the gap from bench to bedside. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Tsiantoulas, D.; Diehl, C.J.; Witztum, J.L.; Binder, C.J. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014, 114, 1743–1756. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; You, B.; Chen, S.P.; Liao, J.K.; Sun, J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: Role of tumor necrosis factor-alpha. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, M.E.; Jain, A.; Matteini, A.; Beamer, B.A.; Wang, N.Y.; Leng, S.X.; Punjabi, N.M.; Walston, J.D.; Fedarko, N.S. Serum levels of the immune activation marker neopterin change with age and gender and are modified by race, BMI, and percentage of body fat. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 858–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murr, C.; Winklhofer-Roob, B.M.; Schroecksnadel, K.; Maritschnegg, M.; Mangge, H.; Bohm, B.O.; Winkelmann, B.R.; Marz, W.; Fuchs, D. Inverse association between serum concentrations of neopterin and antioxidants in patients with and without angiographic coronary artery disease. Atherosclerosis 2009, 202, 543–549. [Google Scholar] [CrossRef]
- Murr, C.; Widner, B.; Wirleitner, B.; Fuchs, D. Neopterin as a marker for immune system activation. Curr. Drug Metab. 2002, 3, 175–187. [Google Scholar] [CrossRef]
- Fuchs, D.; Avanzas, P.; Arroyo-Espliguero, R.; Jenny, M.; Consuegra-Sanchez, L.; Kaski, J.C. The role of neopterin in atherogenesis and cardiovascular risk assessment. Curr. Med. Chem. 2009, 16, 4644–4653. [Google Scholar] [CrossRef]
- Coquery, C.M.; Wade, N.S.; Loo, W.M.; Kinchen, J.M.; Cox, K.M.; Jiang, C.; Tung, K.S.; Erickson, L.D. Neutrophils contribute to excess serum BAFF levels and promote CD4+ T cell and B cell responses in lupus-prone mice. PLoS ONE 2014, 9, e102284. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Ng, L.G. Granulopoiesis and Neutrophil Homeostasis: A Metabolic, Daily Balancing Act. Trends Immunol. 2019, 40, 598–612. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.E.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019, 566, 383–387. [Google Scholar] [CrossRef]
- Gaul, D.S.; Stein, S.; Matter, C.M. Neutrophils in cardiovascular disease. Eur. Heart J. 2017, 38, 1702–1704. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, N.; Cherpokova, D.; Martinod, K.; Wagner, D.D. Extracellular DNA NET-Works With Dire Consequences for Health. Circ. Res. 2019, 125, 470–488. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Shi, H.; Winkler, M.A.; Lee, R.; Weintraub, N.L. Perivascular Adipose Tissue and Vascular Perturbation/Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lu, X.; Shi, G.P. Vasa vasorum in atherosclerosis and clinical significance. Int. J. Mol. Sci. 2015, 16, 11574–11608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, T.; Zhang, B.; Wang, Z.; Joehanes, R.; Zhu, J.; Johnson, A.D.; Ying, S.; Munson, P.J.; Raghavachari, N.; Wang, R.; et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1427–1434. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, N.; Hasselwander, S.; Reifenberg, G.; Habermeier, A.; Closs, E.I.; Mimmler, M.; Jung, R.; Karbach, S.; Lagrange, J.; Wenzel, P.; et al. B Lymphocyte-Deficiency in Mice Causes Vascular Dysfunction by Inducing Neutrophilia. Biomedicines 2021, 9, 1686. https://doi.org/10.3390/biomedicines9111686
Xia N, Hasselwander S, Reifenberg G, Habermeier A, Closs EI, Mimmler M, Jung R, Karbach S, Lagrange J, Wenzel P, et al. B Lymphocyte-Deficiency in Mice Causes Vascular Dysfunction by Inducing Neutrophilia. Biomedicines. 2021; 9(11):1686. https://doi.org/10.3390/biomedicines9111686
Chicago/Turabian StyleXia, Ning, Solveig Hasselwander, Gisela Reifenberg, Alice Habermeier, Ellen I. Closs, Maximilian Mimmler, Rebecca Jung, Susanne Karbach, Jérémy Lagrange, Philip Wenzel, and et al. 2021. "B Lymphocyte-Deficiency in Mice Causes Vascular Dysfunction by Inducing Neutrophilia" Biomedicines 9, no. 11: 1686. https://doi.org/10.3390/biomedicines9111686
APA StyleXia, N., Hasselwander, S., Reifenberg, G., Habermeier, A., Closs, E. I., Mimmler, M., Jung, R., Karbach, S., Lagrange, J., Wenzel, P., Daiber, A., Münzel, T., Hövelmeyer, N., Waisman, A., & Li, H. (2021). B Lymphocyte-Deficiency in Mice Causes Vascular Dysfunction by Inducing Neutrophilia. Biomedicines, 9(11), 1686. https://doi.org/10.3390/biomedicines9111686