
Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective

 ,
,  ,
,  , ,
, , 
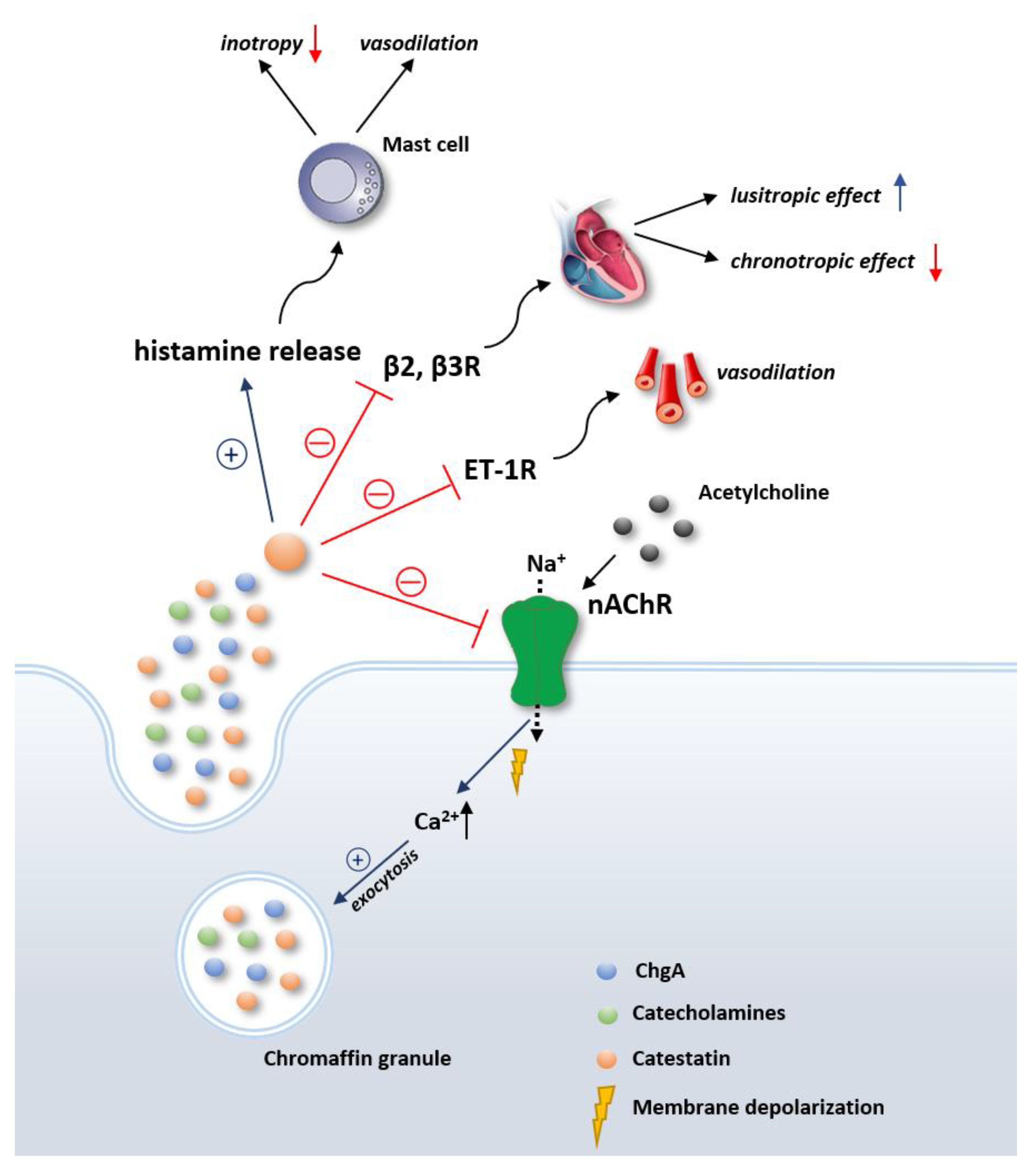{kind=link}
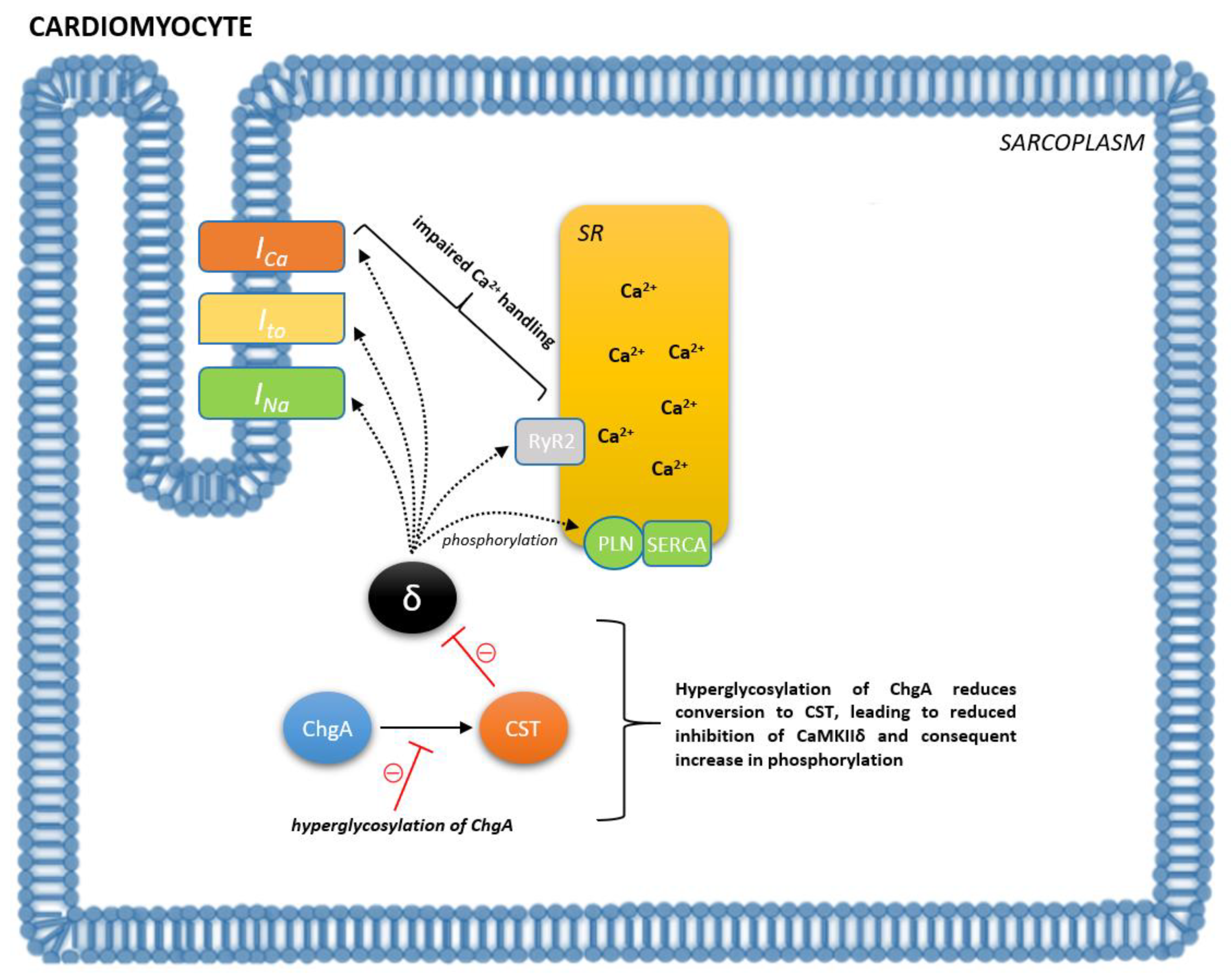{kind=link}
Abstract
:1. Introduction
2. Overview of the Physiological Effects of Catestatin
3. Catestatin in Cardiovascular System Regulation
4. Role of Catestatin in Various Cardiovascular Disorders
5. Biomarker Potential of Catestatin and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Cardiovascular Diseases (CVDs). 2016. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 10 October 2021).
- Packer, M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992, 20, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef] [PubMed]
- de Lucia, C.; Piedepalumbo, M.; Paolisso, G.; Koch, W.J. Sympathetic nervous system in age-related cardiovascular dysfunction: Pathophysiology and therapeutic perspective. Int. J. Biochem. Cell Biol. 2019, 108, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Floras, J.S.; Ponikowski, P. The sympathetic/parasympathetic imbalance in heart failure with reduced ejection fraction. Eur. Heart J. 2015, 36, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Kiuchi, M.G.; Nolde, J.M.; Villacorta, H.; Carnagarin, R.; Chan, J.J.S.; Lugo-Gavidia, L.M.; Ho, J.K.; Matthews, V.B.; Dwivedi, G.; Schlaich, M.P. New Approaches in the Management of Sudden Cardiac Death in Patients with Heart Failure-Targeting the Sympathetic Nervous System. Int. J. Mol. Sci. 2019, 20, 2430. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.E.; Januzzi, J.L. Established and Emerging Roles of Biomarkers in Heart Failure. Circ. Res. 2018, 123, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Mahata, S.K.; O’Connor, D.T.; Mahata, M.; Yoo, S.H.; Taupenot, L.; Wu, H.; Gill, B.M.; Parmer, R.J. Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J. Clin. Investig. 1997, 100, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Mahata, S.K.; Kiranmayi, M.; Mahapatra, N.R. Catestatin: A Master Regulator of Cardiovascular Functions. Curr. Med. Chem. 2018, 25, 1352–1374. [Google Scholar] [CrossRef]
- Biswas, N.; Rodriguez-Flores, J.L.; Courel, M.; Gayen, J.R.; Vaingankar, S.M.; Mahata, M.; Torpey, J.W.; Taupenot, L.; O’Connor, D.T.; Mahata, S.K. Cathepsin L colocalizes with chromogranin a in chromaffin vesicles to generate active peptides. Endocrinology 2009, 150, 3547–3557. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.V.; Hawkins, K.; Jin, L.; Kulig, E.; Fields, K. Chromogranin A, chromogranin B and secretogranin II mRNAs in the pituitary and adrenal glands of various mammals. Regulation of chromogranin A, chromogranin B and secretogranin II mRNA levels by estrogen. Lab. Investig. 1992, 67, 394–404. [Google Scholar]
- Bianco, M.; Gasparri, A.M.; Colombo, B.; Curnis, F.; Girlanda, S.; Ponzoni, M.; Bertilaccio, M.T.; Calcinotto, A.; Sacchi, A.; Ferrero, E.; et al. Chromogranin A Is Preferentially Cleaved into Proangiogenic Peptides in the Bone Marrow of Multiple Myeloma Patients. Cancer Res. 2016, 76, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aardal, S.; Helle, K.B.; Elsayed, S.; Reed, R.K.; Serck-Hanssen, G. Vasostatins, comprising the N-terminal domain of chromogranin A, suppress tension in isolated human blood vessel segments. J. Neuroendocrinol. 1993, 5, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.A.; Glavas, D.; Susilovic Grabovac, Z.; Supe Domic, D.; Stanisic, L.; D’Amario, D.; Duplancic, D.; Bozic, J. Right Ventricular Free Wall Strain and Congestive Hepatopathy in Patients with Acute Worsening of Chronic Heart Failure: A CATSTAT-HF Echo Substudy. J. Clin. Med. 2020, 9, 1317. [Google Scholar] [CrossRef] [PubMed]
- Biswas, N.; Curello, E.; Connor, D.T.O.; Mahata, S.K. Chromogranin/secretogranin proteins in murine heart: Myocardialproduction of chromogranin A fragment catestatin (Chga(364384)). Cell Tissue Res. 2010, 342, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Herrero, C.J.; Alés, E.; Pintado, A.J.; López, M.G.; García-Palomero, E.; Mahata, S.K.; O’Connor, D.T.; García, A.G.; Montiel, C. Modulatory mechanism of the endogenous peptide catestatin on neuronal nicotinic acetylcholine receptors and exocytosis. J. Neurosci. 2002, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.P.; Mahata, S.K.; O’Connor, D.T.; Ziegler, M.G. Mechanism of cardiovascular actions of the chromogranin A fragment catestatin in vivo. Peptides 1998, 19, 1241–1248. [Google Scholar] [CrossRef]
- Mahata, S.K.; Mahata, M.; Parmer, R.J.; OConnor, D.T. Desensitization of catecholamine release. The novel catecholamine release-inhibitory peptide catestatin (chromogranina344364) acts at the receptor to prevent nicotinic cholinergic tolerance. J. Biol. Chem. 1999, 274, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Taupenot, L.; Mahata, S.K.; Mahata, M.; Parmer, R.J.; OConnor, D.T. Interaction of the catecholamine releaseinhibitory peptide catestatin (human chromogranin A (352372)) with the chromaffin cell surface and Torpedo electroplax: Implications for nicotinic cholinergic antagonism. Regul. Pept. 2000, 95, 9–17. [Google Scholar] [CrossRef]
- Helle, K.B.; Corti, A.; Metz-Boutigue, M.H.; Tota, B. The endocrine role for chromogranin A: A prohormone for peptides with regulatory properties. Cell. Mol. Life Sci. 2007, 64, 2863–2886. [Google Scholar] [CrossRef]
- Angelone, T.; Quintieri, A.M.; Brar, B.K.; Limchaiyawat, P.T.; Tota, B.; Mahata, S.K.; Cerra, M.C. The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology 2008, 149, 4780–4793. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Jesmin, S.; Takahashi, Y.; Hatta, E.; Kobayashi, M.; Matsuyama, K.; Kawakami, N.; Sakuma, I.; Gando, S.; Fukui, H.; et al. Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharmacol. Exp. Ther. 2004, 309, 786–795. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Zhang, Z.Y.; Wang, R.X. Protective Mechanisms of Quercetin against Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2020, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Mazza, R.; Gattuso, A.; Mannarino, C.; Brar, B.K.; Barbieri, S.F.; Tota, B.; Mahata, S.K. Catestatin (chromogranin A344364) is a novel cardiosuppressive agent: Inhibition of isoproterenol and endothelin signaling in the frog heart. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H113–H122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, G.; Mahata, S.K.; Cadman, P.; Mahata, M.; Ghosh, S.; Mahapatra, N.R.; Rao, F.; Stridsberg, M.; Smith, D.W.; Mahboubi, P.; et al. Both rare and common polymorphisms contribute functional variation at CHGA, a regulator of catecholamine physiology. Am. J. Hum. Genet. 2004, 74, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Mahata, S.K.; Mahata, M.; Wen, G.; Wong, W.B.; Mahapatra, N.R.; Hamilton, B.A.; OConnor, D.T. The catecholamine release-inhibitory catestatin fragment of chromogranina: Naturally occurring human variants with different potenciesfor multiple chromaffin cell nicotinic cholinergic responses. Mol. Pharmacol. 2004, 66, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borovac, J.A.; Dogas, Z.; Supe-Domic, D.; Galic, T.; Bozic, J. Catestatin serum levels are increased in male patients with obstructive sleep apnea. Sleep Breath 2019, 23, 473–481. [Google Scholar] [CrossRef]
- Widiapradja, A.; Chunduri, P.; Levick, S.P. The role of neuropeptides in adverse myocardial remodeling and heart failure. Cell. Mol. Life Sci. 2017, 74, 2019–2038. [Google Scholar] [CrossRef]
- Gaede, A.H.; Pilowsky, P.M. Catestatin in rat RVLM is sympathoexcitatory, increases barosensitivity, and attenuates chemosensitivity and the somatosympathetic reflex. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1538–R1545. [Google Scholar] [CrossRef] [Green Version]
- Gaede, A.H.; Pilowsky, P.M. Catestatin, a chromogranin A-derived peptide, is sympathoinhibitory and attenuates sympathetic barosensitivity and the chemoreflex in rat CVLM. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R365–R372. [Google Scholar] [CrossRef] [Green Version]
- Avolio, E.; Mahata, S.K.; Mantuano, E.; Mele, M.; Alò, R.; Facciolo, R.M.; Talani, G.; Canonaco, M. Antihypertensive and neuroprotective effects of catestatin in spontaneously hypertensive rats: Interaction with GABAergic transmission in amygdala and brainstem. Neuroscience 2014, 270, 48–57. [Google Scholar] [CrossRef]
- Gayen, J.R.; Gu, Y.; OConnor, D.T.; Mahata, S.K. Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin a null mouse. Endocrinology 2009, 150, 5027–5035. [Google Scholar] [CrossRef]
- Dev, N.B.; Gayen, J.R.; OConnor, D.T.; Mahata, S.K. Chromogranin A and the autonomic system: Decomposition of heart rate variability by time and frequency domains, along with non-linear characteristics during chromogranin A ablation, with rescue by its catestatin. Endocrinology 2010, 151, 2760–2768. [Google Scholar] [CrossRef]
- Rao, F.; Wen, G.; Gayen, J.R.; Das, M.; Vaingankar, S.M.; Rana, B.K.; Mahata, M.; Kennedy, B.P.; Salem, R.M.; Stridsberg, M.; et al. Catecholamine release-inhibitory peptide catestatin (chromogranin A(352–372)): Naturally occurring amino acid variant Gly364Ser causes profound changes in human autonomic activity and alters risk for hypertension. Circulation 2007, 115, 2271–2281. [Google Scholar] [CrossRef] [Green Version]
- Krüger, P.G.; Mahata, S.K.; Helle, K.B. Catestatin (CgA344364) stimulates rat mast cell release of histamine in a manner comparable to mastoparan and other cationic charged neuropeptides. Regul. Pept. 2003, 114, 29–35. [Google Scholar] [CrossRef]
- Fung, M.M.; Salem, R.M.; Mehtani, P.; Thomas, B.; Lu, C.F.; Perez, B.; Rao, F.; Stridsberg, M.; Ziegler, M.G.; Mahata, S.K.; et al. Direct vasoactive effects of the chromogranin A (CHGA) peptide catestatin in humans in vivo. Clin. Exp. Hypertens. 2010, 32, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Shooshtarizadeh, P.; Laventie, B.J.; Colin, D.A.; Chich, J.F.; Vidic, J.; de Barry, J.; Chasserot-Golaz, S.; Delalande, F.; Van Dorsselaer, A.; et al. Two chromogranin a-derived peptides induce calcium entry in human neutrophils by calmodulin-regulated calcium independent phospholipase A2. PLoS ONE 2009, 4, e4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frodermann, V.; Nahrendorf, M. Neutrophil-macrophage cross-talk in acute myocardial infarction. Eur. Heart J. 2017, 38, 198–200. [Google Scholar] [CrossRef]
- Bassino, E.; Fornero, S.; Gallo, M.P.; Gallina, C.; Femminò, S.; Levi, R.; Tota, B.; Alloatti, G. Catestatin exerts direct protective effects on rat cardiomyocytes undergoing ischemia/reperfusion by stimulating PI3K-Akt-GSK3β pathway and preserving mitochondrial membrane potential. PLoS ONE 2015, 10, e0119790. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.Y.; Peng, F.; Wang, J.; Liu, L.; Meng, L.; Zhao, J.; Han, X.N.; Ding, W.H. Catestatin in defense of oxidative-stress-induced apoptosis: A novel mechanism by activating the beta2 adrenergic receptor and PKB/Akt pathway in ischemic-reperfused myocardium. Peptides 2020, 123, 170200. [Google Scholar] [CrossRef]
- Zivkovic, P.M.; Matetic, A.; Tadin Hadjina, I.; Rusic, D.; Vilovic, M.; Supe-Domic, D.; Borovac, J.A.; Mudnic, I.; Tonkic, A.; Bozic, J. Serum Catestatin Levels and Arterial Stiffness Parameters Are Increased in Patients with Inflammatory Bowel Disease. J. Clin. Med. 2020, 9, 628. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Alloatti, G.; Gallo, M.P.; Cerra, M.C.; Levi, R.; Tullio, F.; Bassino, E.; Dolgetta, S.; Mahata, S.K.; Tota, B.; et al. Catestatin improves post-ischemic left ventricular function and decreases ischemia/reperfusion injury in heart. Cell. Mol. Neurobiol. 2010, 30, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumrić, M.; Tičinović Kurir, T.; Borovac, J.A.; Božić, J. The Role of Natural Killer (NK) Cells in Acute Coronary Syndrome: A Comprehensive Review. Biomolecules 2020, 10, 1514. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, M.G.; Tullio, F.; Angotti, C.; Cerra, M.C.; Angelone, T.; Tota, B.; Alloatti, G.; Penna, C.; Pagliaro, P. Catestatin reduces myocardial ischaemia/reperfusion injury: Involvement of PI3K/Akt, PKCs, mitochondrial KATP channels and ROS signalling. Pflugers Arch. 2013, 465, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Zheng, Y.; Cai, J.; Fan, J.; Wang, J.; Yang, J.; Cui, Q.; Xu, G.; Tang, C.; Geng, B. Catestatin attenuates endoplasmic reticulum induced cell apoptosis by activation type 2 muscarinic acetylcholine receptor in cardiac ischemia/reperfusion. Sci. Rep. 2015, 5, 16590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brar, B.K.; Helgeland, E.; Mahata, S.K.; Zhang, K.; O’Connor, D.T.; Helle, K.B.; Jonassen, A.K. Human catestatin peptides differentially regulate infarct size in the ischemic-reperfused rat heart. Regul. Pept. 2010, 165, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahapatra, N.R.; OConnor, D.T.; Vaingankar, S.M.; Hikim, A.P.; Mahata, M.; Ray, S.; Staite, E.; Wu, H.; Gu, Y.; Dalton, N.; et al. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J. Clin. Investig. 2005, 115, 1942–1952. [Google Scholar] [CrossRef] [Green Version]
- Kiranmayi, M.; Chirasani, V.R.; Allu, P.K.; Subramanian, L.; Martelli, E.E.; Sahu, B.S.; Vishnuprabu, D.; Kumaragurubaran, R.; Sharma, S.; Bodhini, D.; et al. Catestatin Gly364Ser Variant Alters Systemic Blood Pressure and the Risk for Hypertension in Human Populations via Endothelial Nitric Oxide Pathway. Hypertension 2016, 68, 334–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, B.S.; Obbineni, J.M.; Sahu, G.; Allu, P.K.; Subramanian, L.; Sonawane, P.J.; Singh, P.K.; Sasi, B.K.; Senapati, S.; Maji, S.K.; et al. Functional genetic variants of the catecholamine-release-inhibitory peptide catestatin in an Indian population: Allele-specific effects on metabolic traits. J. Biol. Chem. 2012, 287, 43840–43852. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, D.T.; Zhu, G.; Rao, F.; Taupenot, L.; Fung, M.M.; Das, M.; Mahata, S.K.; Mahata, M.; Wang, L.; Zhang, K.; et al. Heritability and genome-wide linkage in US and Australian twins identify novel genomic regions controlling Chromogranin A: Implications for secretion and blood pressure. Circulation 2008, 118, 247–257. [Google Scholar] [CrossRef]
- O’Connor, D.T.; Kailasam, M.T.; Kennedy, B.P.; Ziegler, M.G.; Yanaihara, N.; Parmer, R.J. Early decline in the catecholamine release-inhibitory peptide catestatin in humans at genetic risk of hypertension. J. Hypertens. 2002, 20, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.H.; Carlson, C.R.; Louch, W.E.; Dahl, M.B.; Sandbu, R.A.; Johansen, R.F.; Jarstadmarken, H.; Bjørås, M.; Høiseth, A.D.; Brynildsen, J.; et al. Glycosylated Chromogranin A in Heart Failure: Implications for Processing and Cardiomyocyte Calcium Homeostasis. Circ. Heart Fail. 2017, 10, e003675. [Google Scholar] [CrossRef] [Green Version]
- Lunde, I.G.; Aronsen, J.M.; Kvaløy, H.; Qvigstad, E.; Sjaastad, I.; Tønnessen, T.; Christensen, G.; Grønning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Tang, K.; Avolio, E.; Schilling, J.M.; Pasqua, T.; Liu, M.A.; Cheng, H.; Gao, H.; Zhang, J.; Mahata, S.; et al. Immunosuppression of Macrophages Underlies the Cardioprotective Effects of CST (Catestatin). Hypertension 2021, 77, 1670–1682. [Google Scholar] [CrossRef]
- Wang, D.; Liu, T.; Shi, S.; Li, R.; Shan, Y.; Huang, Y.; Hu, D.; Huang, C. Chronic Administration of Catestatin Improves Autonomic Function and Exerts Cardioprotective Effects in Myocardial Infarction Rats. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 526–535. [Google Scholar] [CrossRef]
- Dev, N.B.; Mir, S.A.; Gayen, J.R.; Siddiqui, J.A.; Mustapic, M.; Vaingankar, S.M. Cardiac electrical activity in a genomically “humanized” chromogranin a monogenic mouse model with hyperadrenergic hypertension. J. Cardiovasc. Transl. Res. 2014, 7, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Ma, D.; Ji, L.; Zhang, J.; Su, J.; Xue, W.; Chen, X.; Wang, W. Usefulness of catestatin to predict malignant arrhythmia in patients with acute myocardial infarction. Peptides 2014, 55, 131–135. [Google Scholar] [CrossRef]
- Burchell, A.E.; Sobotka, P.A.; Hart, E.C.; Nightingale, A.K.; Dunlap, M.E. Chemohypersensitivity and autonomic modulation of venous capacitance in the pathophysiology of acute decompensated heart failure. Curr. Heart Fail. Rep. 2013, 10, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Fallick, C.; Sobotka, P.A.; Dunlap, M.E. Sympathetically mediated changes in capacitance: Redistribution of the venous reservoir as a cause of decompensation. Circ. Heart Fail. 2011, 4, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieroni, M.; Corti, A.; Tota, B.; Curnis, F.; Angelone, T.; Colombo, B.; Cerra, M.C.; Bellocci, F.; Crea, F.; Maseri, A. Myocardial production of chromogranin A in human heart: A new regulatory peptide of cardiac function. Eur. Heart J. 2007, 28, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Ceconi, C.; Ferrari, R.; Bachetti, T.; Opasich, C.; Volterrani, M.; Colombo, B.; Parrinello, G.; Corti, A. Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur. Heart J. 2002, 23, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omland, T.; Dickstein, K.; Syversen, U. Association between plasma chromogranin A concentration and long-term mortality after myocardial infarction. Am. J. Med. 2003, 114, 25–30. [Google Scholar] [CrossRef]
- Estensen, M.E.; Hognestad, A.; Syversen, U.; Squire, I.; Ng, L.; Kjekshus, J.; Dickstein, K.; Omland, T. Prognostic value of plasma chromogranin A levels in patients with complicated myocardial infarction. Am. Heart J. 2006, 152, 927.e1–927.e6. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.K.; Mahata, S.K. Chromogranin A Regulation of Obesity and Peripheral Insulin Sensitivity. Front. Endocrinol. 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, A.M.; Røsjø, H.; Omland, T.; Karlsson, T.; Hartford, M.; Flyvbjerg, A.; Caidahl, K. Prognostic value of circulating chromogranin A levels in acute coronary syndromes. Eur. Heart J. 2009, 30, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Røsjø, H.; Masson, S.; Latini, R.; Flyvbjerg, A.; Milani, V.; La Rovere, M.T.; Revera, M.; Mezzani, A.; Tognoni, G.; Tavazzi, L.; et al. Prognostic value of chromogranin A in chronic heart failure: Data from the GISSI-Heart Failure trial. Eur. J. Heart Fail. 2010, 12, 549–556. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, F.; Yu, H.; Mi, L.; Gao, W. Catestatin is useful in detecting patients with stage B heart failure. Biomarkers 2011, 16, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ding, W.; Li, R.; Ye, X.; Zhao, J.; Jiang, J.; Meng, L.; Wang, J.; Chu, S.; Han, X.; et al. Plasma levels and diagnostic value of catestatin in patients with heart failure. Peptides 2013, 46, 20–25. [Google Scholar] [CrossRef]
- Borovac, J.A.; Glavas, D.; Susilovic Grabovac, Z.; Supe Domic, D.; D’Amario, D.; Bozic, J. Catestatin in Acutely Decompensated Heart Failure Patients: Insights from the CATSTAT-HF Study. J. Clin. Med. 2019, 8, 1132. [Google Scholar] [CrossRef] [Green Version]
- Mazza, R.; Tota, B.; Gattuso, A. Cardio-vascular activity of catestatin: Interlocking the puzzle pieces. Curr. Med. Chem. 2015, 22, 292–304. [Google Scholar] [CrossRef]
- Alam, M.J.; Gupta, R.; Mahapatra, N.R.; Goswami, S.K. Catestatin reverses the hypertrophic effects of norepinephrine in H9c2 cardiac myoblasts by modulating the adrenergic signaling. Mol. Cell. Biochem. 2020, 464, 205–219. [Google Scholar] [CrossRef]
- Meng, L.; Ye, X.J.; Ding, W.H.; Yang, Y.; Di, B.B.; Liu, L.; Huo, Y. Plasma catecholamine release-inhibitory peptide catestatin in patients with essential hypertension. J. Cardiovasc. Med. 2011, 12, 643–647. [Google Scholar] [CrossRef]
- Peng, F.; Chu, S.; Ding, W.; Liu, L.; Zhao, J.; Cui, X.; Li, R.; Wang, J. The predictive value of plasma catestatin for all-cause and cardiac deaths in chronic heart failure patients. Peptides 2016, 86, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wołowiec, Ł.; Rogowicz, D.; Banach, J.; Gilewski, W.; Sinkiewicz, W.; Grześk, G. Catestatin as a New Prognostic Marker in Stable Patients with Heart Failure with Reduced Ejection Fraction in Two-Year Follow-Up. Dis. Markers 2020, 2020, 8847211. [Google Scholar] [CrossRef]
- Izci, S.; Acar, E.; Inanir, M. Plasma catestatin level predicts sPESI score and mortality in acute pulmonary embolism. Arch. Med. Sci. Atheroscler. Dis. 2020, 5, e49–e56. [Google Scholar] [CrossRef]
- Chan, C.M.; Woods, C.; Shorr, A.F. The validation and reproducibility of the pulmonary embolism severity index. J. Thromb. Haemost. 2010, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Bourebaba, Y.; Mularczyk, M.; Marycz, K.; Bourebaba, L. Catestatin peptide of chromogranin A as a potential new target for several risk factors management in the course of metabolic syndrome. Biomed. Pharmacother. 2021, 134, 111113. [Google Scholar] [CrossRef]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase to Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Mahata, S.; Bandyopadhyay, G.K.; Zhou, Z.; Wollam, J.; Vu, J.; Mayoral, R.; Chi, N.W.; Webster, N.J.G.; Corti, A.; et al. Catestatin Inhibits Obesity-Induced Macrophage Infiltration and Inflammation in the Liver and Suppresses Hepatic Glucose Production, Leading to Improved Insulin Sensitivity. Diabetes 2018, 67, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Bandyopadhyay, G.K.; Ray, I.; Bandyopadhyay, K.; Chowdhury, N.; De, R.K.; Mahata, S.K. Catestatin improves insulin sensitivity by attenuating endoplasmic reticulum stress: In vivo and in silico validation. Comput. Struct. Biotechnol. J. 2020, 18, 464–481. [Google Scholar] [CrossRef]
- Simunovic, M.; Supe-Domic, D.; Karin, Z.; Degoricija, M.; Paradzik, M.; Bozic, J.; Unic, I.; Skrabic, V. Serum catestatin concentrations are decreased in obese children and adolescents. Pediatr. Diabetes 2019, 20, 549–555. [Google Scholar] [CrossRef]
- Luketin, M.; Mizdrak, M.; Boric-Skaro, D.; Martinovic, D.; Tokic, D.; Vilovic, M.; Supe-Domic, D.; Ticinovic Kurir, T.; Bozic, J. Plasma Catestatin Levels and Advanced Glycation End Products in Patients on Hemodialysis. Biomolecules 2021, 11, 456. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozic, J.; Kumric, M.; Ticinovic Kurir, T.; Urlic, H.; Martinovic, D.; Vilovic, M.; Tomasovic Mrcela, N.; Borovac, J.A. Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective. Biomedicines 2021, 9, 1757. https://doi.org/10.3390/biomedicines9121757
Bozic J, Kumric M, Ticinovic Kurir T, Urlic H, Martinovic D, Vilovic M, Tomasovic Mrcela N, Borovac JA. Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective. Biomedicines. 2021; 9(12):1757. https://doi.org/10.3390/biomedicines9121757
Chicago/Turabian StyleBozic, Josko, Marko Kumric, Tina Ticinovic Kurir, Hrvoje Urlic, Dinko Martinovic, Marino Vilovic, Nada Tomasovic Mrcela, and Josip A. Borovac. 2021. "Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective" Biomedicines 9, no. 12: 1757. https://doi.org/10.3390/biomedicines9121757
APA StyleBozic, J., Kumric, M., Ticinovic Kurir, T., Urlic, H., Martinovic, D., Vilovic, M., Tomasovic Mrcela, N., & Borovac, J. A. (2021). Catestatin as a Biomarker of Cardiovascular Diseases: A Clinical Perspective. Biomedicines, 9(12), 1757. https://doi.org/10.3390/biomedicines9121757