1. Introduction
7,7″-Dimethoxyagastisflavone (DMGF) is a biflavonoid isolated from the needles of
Taxus media var. Hicksii [
1]. The biological activity of DMGF was first revealed when it was found to be an inhibitor of the production of aflatoxin by
Aspergillus flavus [
2]. Our research further demonstrated that DMGF could induce apoptotic and autophagic cell death in various types of cancer cells [
1]. DMGF significantly suppressed the metastatic behaviors of highly invasive melanoma cancer cells, including the intravasation, colonization, and invasion of the lymphatic duct [
3]. DMGF can downregulate the levels of key modulators of the Cdc42/Rac1 pathway to interfere in F-actin polymerization and suppress the formation of lamellipodia by reducing the phosphorylation of CREB [
3]. These findings suggest that DMGF may be further developed as a chemotherapeutic drug for patients with metastatic melanoma; however, the anti-inflammatory activity of DMGF has hitherto not been investigated.
Inflammation is a protective immune response to harmful stimuli, such as pathogens or viruses, driven by the evolutionarily conserved innate immune system. However, an acute systemic inflammatory response such as sepsis usually results in a dysregulated cytokine storm which causes a massive inflammatory cascade and irreversible organ dysfunction [
4]. The clinical treatment of sepsis focuses on treating the primary triggering condition to disrupt the progression of the continuum of septic shock and multi-organ dysfunction syndrome [
4]. After ensuring hemodynamic stability, broad-spectrum antibiotics and glucocorticoids are used to improve survival and the reversal of shock in patients [
4,
5]. Glucocorticoid treatment can counteract several sepsis characteristics, including excessive inflammation, vascular defects, and hypoglycemia [
6]. However, glucocorticoid resistance can cause hypothalamic–pituitary–adrenal axis dysfunction, which hampers the maintenance of homeostasis and regulation of cytokines [
6]. The management of systemic inflammatory responses is expected to improve the treatment of sepsis in the future.
During the inflammatory response, proinflammatory cytokines and chemokines increase immune cell recruitment. The recruitment of immune cells to the infection site requires their trafficking across the blood vessel wall [
7]. Under inflammatory conditions, inflammatory monocytes/macrophages transmigrate following a series of events, including rolling adhesion, tight binding, diapedesis (the squeezing of cells through blood cell walls), and migration [
7]. Rolling adhesion on vascular surfaces is the first step in recruiting circulating leukocytes, hematopoietic progenitors, or platelets to specific organs or to sites of infection or injury. Rolling requires the rapid yet balanced formation and dissociation of adhesive bonds in the challenging environment of blood flow. Initially, proinflammatory cytokines enhance the expression of adhesion molecules, such as P-selectin, E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) on the surface of the endothelial cells [
7]. These adhesion molecules expressed on endothelial cells are required for cell migration. Inflammatory monocytes express a high level of P-selectin glycoprotein ligand-1 (PSGL-1), which transiently interacts with P- and E-selectin and mediates rolling on the vascular endothelium [
8]. The increased expression of PSGL-1 has been found to be associated with inflammation [
9]. Monocyte/macrophage rolling strongly depends on monocyte-expressed very late antigen-4 (VLA-4 α4β1 integrin), which binds to VCAM-1 and allows slow rolling on the cytokine-activated endothelium, thereby promoting the transition between rolling and tight binding. Monocyte rolling and arrest also depend on the leucocyte integrin lymphocyte function-associated antigen-1 (LFA-1) and macrophage-1-antigen (Mac-1), which bind to endothelial ICAM-1 and ICAM-2. When exposed to C-C and C-X-C chemokines, integrins expressed on the surfaces of monocytes tightly bind to adhesion molecules on the endothelium. The proinflammatory mediators have been reported to prime leukocytes for integrin expression and activation [
9]. Increased LFA-1 levels on monocytes are associated with virus infection [
10]. Finally, monocytes squeeze between the endothelial cells (diapedesis), penetrate the connective tissue, and move to the infection site. In the diapedesis stage, the distribution of adhesion molecules and actin cytoskeleton of monocytes shows dynamic changes [
11]. Therefore, when macrophages receive inflammatory stimuli, they release cytokines and chemokines to promote the recruitment of more inflammatory cells. The recruitment of these cells to the inflammation site requires their movement through rolling and adhesion in the blood vessels. Based on these factors, the effective inhibition of cytokine and chemokine release and macrophage migration are the important factors that inhibit the inflammatory response.
In this study, the anti-inflammatory effects of DMGF were investigated. The in vitro results indicated that DMGF efficiently decreased the release of LPS-induced proinflammatory cytokines and other chemo-mediators. DMGF also inhibited inflammatory macrophage migration through downregulating the expression of adhesion molecules and the assembly of F-actin. In a mouse model of endotoxin shock, DMGF not only decreased the expression of proinflammatory cytokines in sera but also decreased the infiltration of macrophages. DMGF reversed LPS-induced tissue damage and maintained the functions of various organs. Furthermore, DMGF downregulated the transcriptional activity of NF-κB and estrogen receptor alpha (ERα), thus reducing cytokine release and transmigration in inflammatory macrophages.
2. Materials and Methods
2.1. DMGF Preparation
DMGF (98% purity by high-performance liquid chromatography) was isolated from the dried needles of
Taxus media var. Hicksii and purified according to the method described previously [
1,
3].
2.2. Cells and Cell Cultures
RAW264.7 mouse macrophages were purchased from the Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan, ROC) and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco/Invitrogen, Carlsbad, CA, USA) supplemented with heat-inactivated 10% fetal bovine serum (Gibco/Invitrogen) and 1% Penicillin/streptomycin (Gibco/Invitrogen) in 5% CO2 at 37 °C. Mouse splenocytes were isolated from BALB/c mice according to the following procedures. Briefly, BALB/c mice were sacrificed and their spleens were collected. The spleen was placed into the cell strainer and homogenized through the cell strainer into the Petri dish. Erythrocytes in the homogenized spleen were lysed by ACK lysis buffer at room temperature for 5 min following the PBS wash. After the spinning down of the cells, they were resuspended and cultured with RPMI1640 growth medium (Gibco/Invitrogen) supplemented with heat-inactivated 10% fetal bovine serum (Gibco/Invitrogen) and 1% Penicillin/streptomycin (Gibco/Invitrogen).
2.3. Cell Proliferation Assay
Cells were seeded at 1 × 104 cells/well overnight in a 96-well microculture plate. After treatment with serial concentrations of DMGF in dimethyl sulfoxide (DMSO, final concentration of DMSO is 0.1%) for 48 h, their cell proliferation was measured by CCK-8 (Dojindo Molecular Technologies, Kumamoto, Japan) cell counting regents. The cell viability ratio (%) was calculated using the following equation: % viability = absorbance of test sample/absorbance of control × 100%. The results were expressed in duplicate with three independent experiments.
2.4. Proinflammatory Cytokines
Splenocytes were seeded at 1 × 106 cells/well in a 24-well culture plate and then treated with 1 μg/mL LPS and various doses of DMGF for 48 h. The levels of TNF-α, IL-1β, and IL-6 in the culture media were determined by the ELISA kit (R&D System, Minneapolis, MN, USA) according to the manufacturer’s instructions.
2.5. Cytokine Profile Analysis
Splenocytes were seeded at 1 × 106 cells/well in a 24-well culture plate and then treated with 1 μg/mL LPS and 0.6 μg/mL DMGF for 48 h. The supernatants in each group were collected for analysis of cytokine levels by a cytokine multiplex assay using the Bio-Plex Pro Mouse Cytokine Standard Group I 23-Plex, following the manufacturer’s protocol (Bio-Rad, Hercules, CA, USA).
2.6. Macrophage Migration Assay
Macrophage migration was performed by the Boyden chamber technology. Briefly, RAW264.7 cells were treated with 1 μg/mL LPS and/or 0.6 μg/mL DMGF and then plated (20,000 cells/well) in the upper well of a transwell plate (5-μm pore; Merck Millipore, Billerica, MA, USA). The cells were allowed to migrate toward the lower well with DMEM growth medium for 10 h. The cells on the top of the filter were removed with a cotton swab and migrated cells on the underside were fixed with methanol, stained with 50 μg/mL propidium iodide and counted with a fluorescence microscope.
2.7. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RAW264.7 cells (3 × 105 cells/well) were treated with 1 μg/mL LPS and 0.6 μg/mL DMGF for 10 h. Then, total cellular RNA was extracted with TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA) and reverse-transcribed into cDNA using the Superscript-III kit (Invitrogen). PCR analysis was performed on aliquots of the cDNA preparations to detect gene expression. The cDNA of Itgal, Itga4, Itgb1, Itgb2, Cdc42, Pak1, n-Wasp, Rac1, Limk, and Esr1 were then amplified by PCR. The primers for mouse Itgal were: forward primer—5′-CATGGGCCACGTTCTCATCT-3′ and reverse primer—5′-GCCAGTCAGCCTACATGGTT-3′; the primers for mouse Itga4 were: forward primer—5′-CAGCAAAAAGGCATAGCGGG-3′ and reverse primer—5′-AGC TCTGTGAGCCATCGAAC-3′; the primers for mouse Itgb1 were: forward primer—5′-ATTGCCAGACGGAGTAACAA-3′ and reverse primer—5′-CTGTGCTACATTCACAGTGC -3′; the primers for mouse Itgb2 were: forward primer—5′-CGGTTTCTTTCCGCCATTAT-3′ and reverse primer—5′-CAAAAGACTCCACAAAGCTTAC-3′; the primers for mouse Cdc42 were: forward primer—5′-CGGAGAAGCTGAGGACAAGAT-3′ and reverse primer—5′-GAGTGTATGGCT CTCCACCAA-3′; the primers for mouse Pak1 were: forward primer—5′-ACTATG ATTGGAGCCGGCAG-3′ and reverse primer—5′-TGGCATTCCCGTAAACTCCC-3′; the primers for mouse n-Wasp were: forward primer—5′-AATGCTGGACACTGG CTACC-3′ and reverse primer—5′-ACGGGGTGGGAGTGAGATAA-3′; the primers for mouse Rac1 were: forward primer—5′- AGATGCAGGCCATCAAGTGT-3′ and reverse primer—5′-TAGGAGAGGGGACGCAATCT-3′; the primers for mouse Limk were: forward primer—5′-CCATCAAGGTGACACACCGA-3′ and reverse primer—5′-GCAAAGCTGACCCTCTGACT-3′; the primers for mouse Esr1 were: forward primer—5′-GCTGAACCGCCCATGATCTA-3′ and reverse primer—5′-CCAGGAGCAAGTTAGGAGCA -3′. All PCR reagents used to amplify the cDNA were purchased from Promega (Madison, WI, USA). Actin cDNA in the samples was used to normalize the loading amounts in each reaction. Finally, PCR products were resolved by electrophoresis on 2% agarose gels, stained with ethidium bromide and photographed using the BioDoc-It Imaging System (UVP, Upland, CA, USA).
2.8. Confocal Immunofluorescence
To observe the expression of LFA-1 and VLA-4, RAW264.7 cells plated on the cover slides were incubated at 37 °C overnight and then treated with 1 μg/mL LPS and 0.6 μg/mL DMGF for 12 h. Cells were fixed with 4% formaldehyde for 10 min and stained by Concanavalin A (Con A) conjugated with Alexa Fluor 488 for cell membrane staining. Then, the cells were, respectively, stained with APC conjugated anti-mouse LFA-1 (Biolegend, San Diego, CA, USA) and Alexa Fluor 647 conjugated anti-mouse VLA-4 (Biolegend) antibodies for 60 min. To observe the structures of lamellipodia, the fixed cells were stained with phallotoxin for 60 min for F-actin staining. The stained cells were observed with DAPI (Vectashie mounting medium, Vector Laboratories, Burlingame, CA, USA) by a Zeiss LSM 780 Confocal microscope (Zeiss, Jena, Germany).
2.9. Western Blot
Cells were treated with 1 μg/mL LPS and 0.6 μg/mL DMGF for 10 h and then lysed in RIPA lysis buffer. The protein concentration of the cell lysate was estimated with the Bradford protein assay using BSA as the standard. Total proteins (50 μg) were separated by SDS-PAGE using a 10% polyacrylamide gel and then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% skim milk in phosphate-buffered saline with Tween 20 (0.05% v/v Tween-20 in PBS, pH 7.2) for 1 h. The membranes were then incubated with primary antibodies such as anti-mouse NF-κB (1:1000, Cell Signaling), anti-mouse IκB (1:1000, Cell Signaling) and anti-mouse ERα antibodies (1:1000, Bioss) at 4 °C overnight, followed by incubation with a horseradish peroxide-linked secondary antibody (1:10,000). The protein bands were visualized using the Amersham ECL Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, UK). The intensity of the chemiluminescence signal was quantified using the Biospectrum 815 Imaging System and Vision Works Software (UVP, Upland, CA, USA).
2.10. NF-κB-GFP Transcriptional Reporter Assay
Due to the low transfection efficiency in RAW264.7 cells (approximately 20%), HEK293 cells were used as the experimental cell model for the NF-κB transcriptional reporter assay. HEK293 cells were seeded at 3 × 105 cells per well and cultured overnight in six-well cultured plates. In transfection assays, 200 μL/well of the transfection sample containing 3 μg plasmid DNA containing NF-κB response element and GFP reporter and 10 μL Lipofectamine (Invitrogen, NY, USA) in Opti-MEM (Invitrogen) were added and incubated with the cells. The 2 mL/well serum containing DMEM complete media was added after 16 h, and the transfected cells were resuspended and divided equally into each well of a 24-well plate. Then, 50 ng/mL of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO, USA) was added for NF-κB induction and 0.6 μg/mL DMGF was administered for 24 h NF-κB–GFP reporter gene expression was assayed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
2.11. Analysis of Cell Surface ER-α Expression by Flow Cytometry
RAW264.7 cells were seeded at 1 × 105 cells/well in a 24-well culture plate overnight and then cells were treated with 1 μg/mL LPS and/or 0.6 μg/mL DMGF. To evaluate ER-α expression on the membrane, cells were harvested, fixed and incubated with anti-mouse ER-α antibody (1:200, GeneTex, Hsinchu, Taiwan, ROC) for 1 h and then the FITC-conjugated secondary antibodies (1:200, Jackson ImmunoResearch, West Grove, PA, USA) for 30 min at 4 °C. Fluorescence related to immunolabeling was analyzed by flow cytometry (BD Biosciences).
2.12. ER-α Transcriptional Reporter Assay
HEK293 cells were seeded at 3 × 104 cells/well in a 96-well culture plate overnight and then co-transfected with pBIND-ER-α and pGL4.35 ERE-Firefly luciferase reporter plasmid (Promega). The vector of pGL4 reporter lacking ERE served as a negative control vector. The transfection efficiency was served by pBIND-ER-α containing the gene of the hRluc-neomycin fusion protein. Cells were harvested in reporter lysis buffer 24 h post-transfection, and lysates were assayed for luciferase activity with a dual luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Luciferase activity was normalized with the ratio of firefly to Renilla luciferase activities.
2.13. Drug Docking
The protein target candidates for DMGF docking were predicted by the SwissTargetPrediction Website (
http://www.swisstargetprediction.ch/, accessed on 24 June 2019). DMGF (red) was docked with estrogen receptor α (ER-α) by iGEMDOCK v2.1 software (
http://gemdock.life.nctu.edu.tw/dock/igemdock.php). ER-α (PBD code: 1QKU) was selected from the Protein Data Bank (PDB). The parameters of iGEMDOCK were set as follows: population—500; generation—70; and solution—100.
2.14. ESR1 Knockdown
Lentiviral particles with the lentiviral vector expressing Esr1 siRNA and scrambled siRNA were purchased from the RNA Technology Platform and Gene Manipulation Core (Academia Sinica, Taipei, Taiwan). The target sequence of Esr1 is 5′- GCCGAAATGAAATGGGTGCTT-3′ (siESR1). The scrambled siRNA sequence is 5′-CCTAAGGTTAAGTCGCCCTCG-3. RAW264.7 cells were subcultured at 5 × 105 cells/well into six-well tissue culture plates overnight. Cells were infected with lentiviral particles at a multiplicity of infection (MOI) of 10. To detect the interference effects of different target, ESR1 mRNA expression was determined using RT-PCR.
2.15. Animals
Female BALB/c mice, 5 weeks of age, were purchased from the National Laboratory Animal Center (Taipei, Taiwan). All studies were performed in the Laboratory Animal Core Facility of Academia Sinica following the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Academia Sinica (19-03-1294).
2.16. Endotoxic Mouse Model
Balb/c mice (5-week-old females) were randomized to receive either 10 mg/kg of DMGF or a saline/DMSO solvent by intraperitoneal (i.p.) injection. Immediately afterward, the mice were i.p. injected with 5 mg/kg of LPS. After treatment for 2, 4, 6 and 8 h, blood samples were harvested from submandibular bleeding and centrifuged at 3000× g for 5 min to obtain the sera. The murine proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6 were measured according the instructions included with the ELISA kits (R&D System, Minneapolis, MN, USA).
2.17. Tissue Damage and Macrophage Infiltration
To investigate the effects of DMGF on suppressing inflammation-induced organ damage, tissue morphology was observed. After sacrificing the DMGF-treated mice, the organs, including the heart, lungs, liver, spleen, and kidneys, were removed and fixed in 10% buffered formalin−saline overnight and then embedded in paraffin blocks. Tissue sections were prepared and stained with hematoxylin and eosin (H&E). The morphology was observed under the Zeiss Axiovert 200M microscope (Zeiss, Oberkochen, Germany).
The infiltrating macrophages in organs were analyzed by immunohistochemistry staining. Tissue sections were stained with anti-F4/80 (1:1000) antibodies for macrophage detection.
2.18. Statistical Analysis
The results were expressed as mean ± SD and analyzed using the SAS statistical software package (SAS Institute, Cary, NC, USA). The t-test was used when comparing two independent samples and the ANOVA test was used when comparing multiple samples. Differences with a p value of less than 0.05 were considered statistically significant.
4. Discussion
DMGF, a bioactive biflavonoid, has anti-tumor and anti-metastatic activities in melanoma cells [
3]. However, little is known about its other functions such as in the inhibition of inflammation except for its effect on inducing tumor death through apoptosis or autophagy in cancer cells [
1]. In vitro and in vivo studies showed that DMGF effectively inhibited inflammation and protected the endotoxin shock mice from hyperinflammation and organ damage caused by inflammatory macrophage infiltration (
Figure 1A and
Figure 6). DMGF significantly decreased the release of pro-inflammatory cytokines and chemokines (
Table 1 and
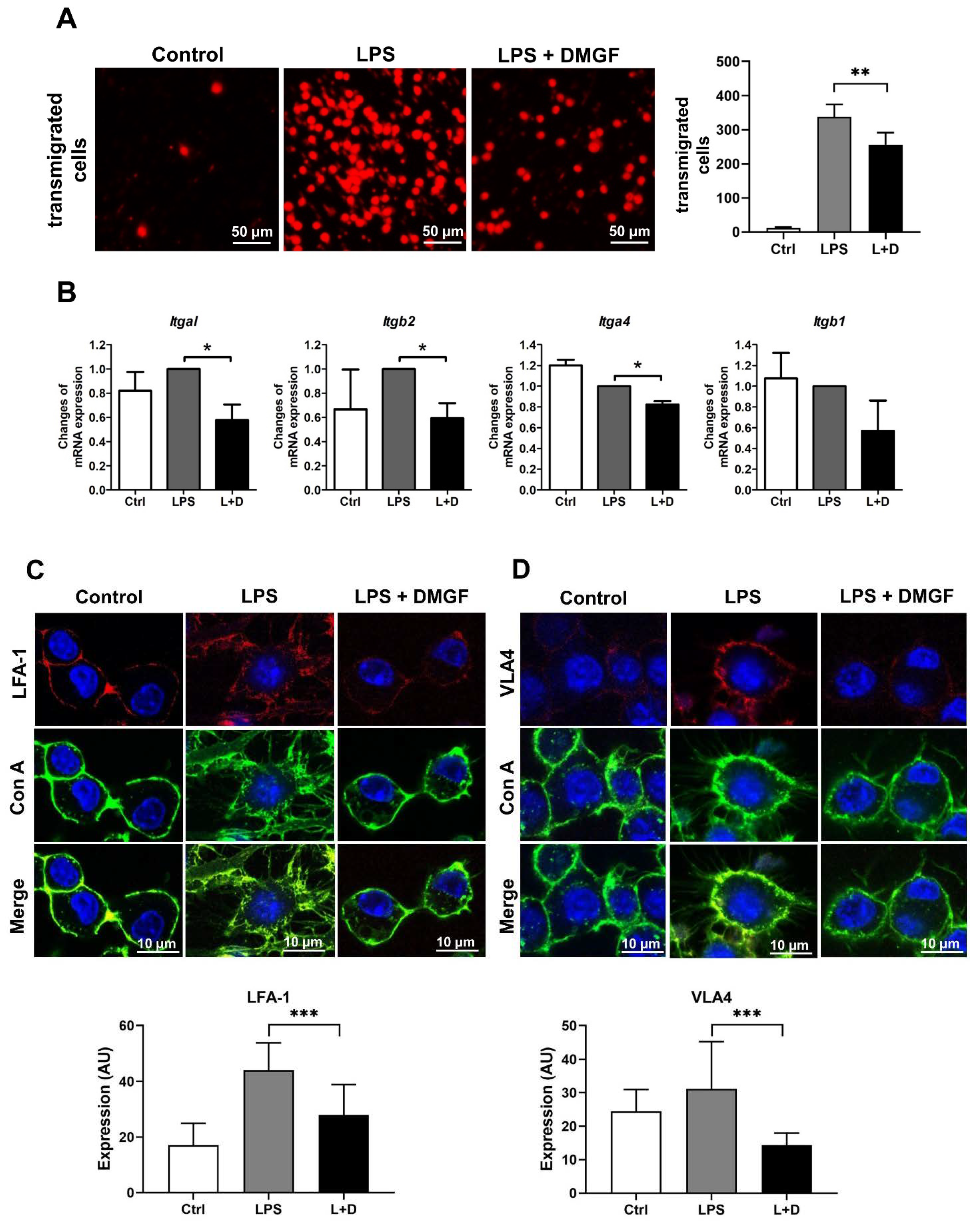
Figure 1B) and reduced the motility of LPS-stimulated macrophages through suppressing adhesion molecules and disrupting actin polymerization (
Figure 2 and
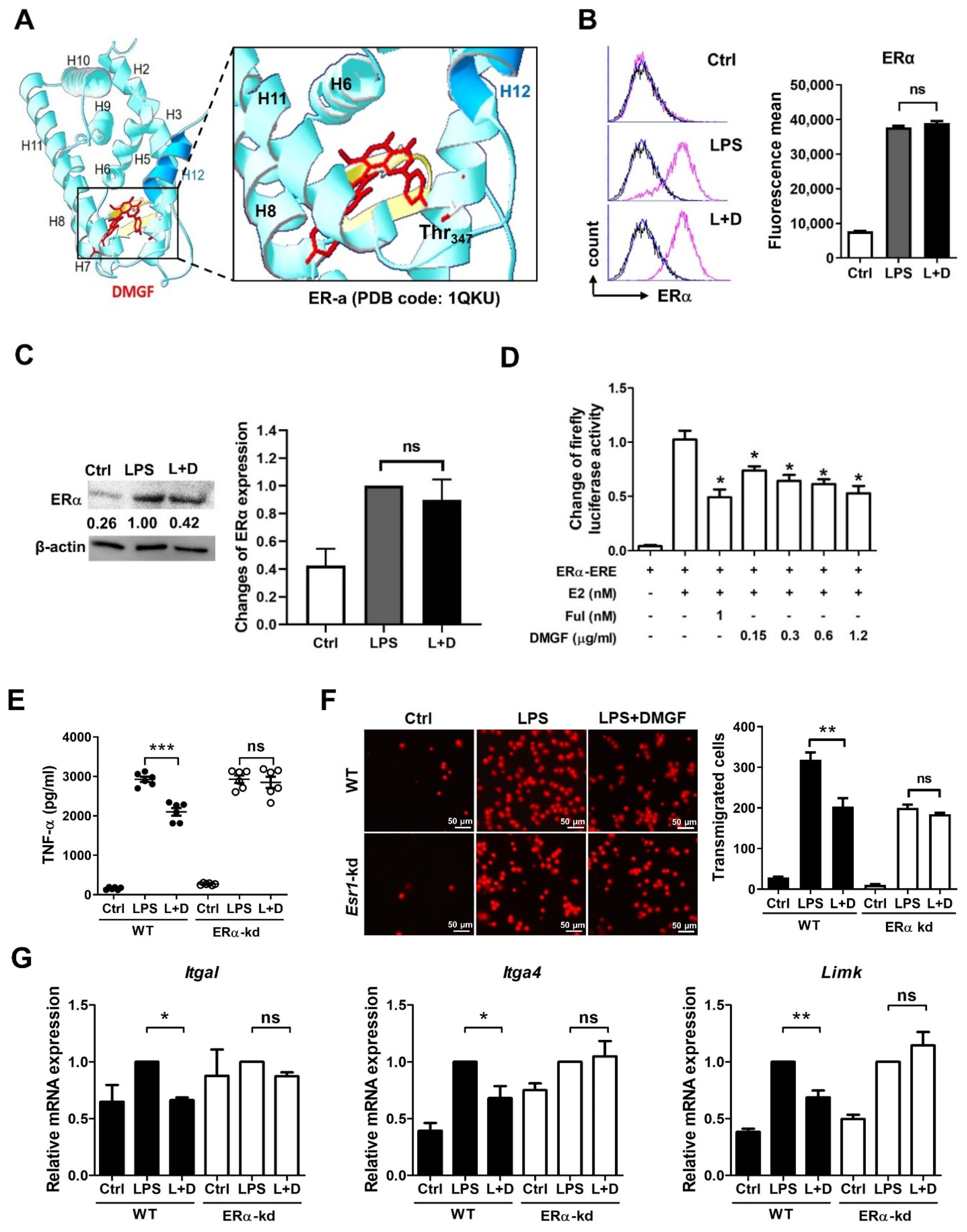
Figure 3). Furthermore, DMGF interacted with the LBD of ERα and downregulated ERα transcriptional activity (
Figure 5A,C). This study suggests the potential of DMGF in the suppression of inflammation through inhibiting the release of pro-inflammatory cytokines and chemokines and interrupting the recruitment of inflammatory cells.
It has been reported that ERα regulates cell signaling pathways in innate and adaptive immunity. ERα is expressed in monocytes/macrophages. In vivo estrogen stimulation promotes the production of proinflammatory cytokines in TLR4 signaling activation of macrophages [
12,
13,
14], while in vitro experiments have proved that estrogen exerts anti-inflammatory properties on monocyte/macrophage cells [
15]. Recently, selective ER modulator drugs such as tamoxifen, raloxifene, and some synthetic compounds have been found to reduce inflammation in systemic or tissue-specific inflammatory diseases [
16,
17]. DMGF interacted with the ERα-LBD at Thr347 and repressed the ERα transcriptional activity (
Figure 5A,C) to decrease proinflammatory cytokines and chemokines (
Table 1). Thr347 is a key residue for agonists and antagonists binding in the LBD of ERα. The crystal structure-based and computer-aided studies of human ERα show that the various agonists and antagonists, as well as E2 bound to the ERα LBP via hydrogen bonds with Thr347, provide the high ligand-binding affinity [
18,
19]. In vitro and in vivo results consistently showed that DMGF treatment has an anti-inflammatory effect. DMGF might act as a selective ER modulator for inhibition of LPS-induced inflammation. DMGF not only suppressed the production of inflammatory cytokines but also inhibited cell migration in LPS-stimulated macrophages (
Figure 2A). DMGF effectively inhibited the expression of integrins for cell adhesion and molecules for F-actin polymerization/depolymerization through inhibiting ERα transcription. These regulators have ESR1 transcription factor binding motif on their promoter (
Table S1). DMGF impeded ERα transcriptional activity and provided anti-inflammatory and anti-migratory activity in macrophages.
In addition to ERα, NF-κB is a critical mediator of pro-inflammatory gene induction and regulates innate and adaptive immunity. The canonical NF-κB pathway is responsible for the transcriptional induction of inflammatory genes including TNF-α, IL-1β, and IL-6 [
20]. DMGF effectively inhibited the NF-κB pathway and transcriptional activity to suppress inflammation (
Figure 4A,B). NF-κB signaling can be affected by ERα mediation [
21,
22]. Estrogen inhibits the LPS-induced pro-inflammatory response, resulting in the inhibition of NF-κB transcriptional activity [
15]. Estrogen-bound ERα has been reported to inhibit NF-κB activity through increasing the expression of IκB and decreasing the phosphorylation of IκB [
22]. Furthermore, ER can directly bond to NF-κB and interact with a co-activator to inhibit the transactivation. In this study, DMGF treatment increased IκB expression (
Figure 4A) and inhibited NF-κB transactivation (
Figure 4B). In the virtual analysis, DMGF directly bound to the LBD of ERα (
Figure 5A). These results indicate that DMGF has a similar effect to estrogen or selective ER modulators in terms of blocking the NF-κB transcription. Such inhibitory effects of DMGF on crosstalk between NF-κB and ERα might also be the reason for DMGF’s anti-inflammatory effect.
DMGF is a biflavonoid that belongs to the polyphenol family that is known to have anti-inflammatory and immunomodulatory activity. Recently, a few natural biflavonoids have been discovered from natural plants. A well-known biflavonoid amentoflavone has anti-inflammatory and antioxidative functions [
23]. Amentoflavone inhibits phytohaemagglutinin (PHA) or the LPS-induced production of NO and ROS and release of proinflammatory cytokines including IL-1β, IL-6, TNF-α, and PGE2 [
23]. Amentoflavone exhibits anti-inflammatory activity through the modulation of the ERK signaling pathway [
24]. Theaflavin and its derivatives from black tea block NO synthesis by inhibiting NF-κB activity, thereby reducing LPS-induced inflammation [
25]. Another biflavonoid composed of two molecular kaempferols isolated from the shell of
Camellia oleifera shows strong free radical scavenging activity in vivo and has an analgesic effect on inflammation-induced pain [
26]. DMGF impacts pro-inflammatory cytokine release (
Table 1), the expression of adhesion molecules (
Figure 2) and the regulation of cytoskeleton filaments (
Figure 3) through downregulating NF-κB and ERα transcriptional activity (
Figure 4 and
Figure 5). DMGF effectively prevents the cytokine disorder, inflammatory cell infiltration, and tissue damage caused by LPS-stimulated acute inflammation in endotoxin shock mice (
Figure 6). Therefore, DMGF modulates the process of inflammation through the reduction in the ERα signaling pathway.
The inflammatory effects of several selective ER modulators, such as fulvestrant, tamoxifen, and raloxifene, have been proved [
27,
28,
29]. In our results, fulvestrant inhibited NF-κB expression in the LPS-induced splenocytes (
Figure S3A). However, fulvestrant did not significantly inhibit the production of proinflammatory cytokines, such as TNF -α and IL-6 (
Figure S3B). Our cytokine results were similar to the results in LPS-stimulated microglia. Fulvestrant significantly decreased NO release but did not affect IL-6 release in LPS-stimulated microglia. Even though tamoxifen and raloxifene can decrease the production of NO and proinflammatory cytokines, their actions on cell mobility are not mentioned [
27]. In our results, DMGF alleviated LPS-induced inflammatory responses and tissue damages in endotoxic mice. The infiltration of macrophages in organs is also greatly reduced by DMGF. It is attributed to the ability of DMGF to inhibit LPS-induced inflammation and cell migration.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}