
α-Synuclein Overexpression Increases Dopamine D2/3 Receptor Binding and Immune Activation in a Model of Early Parkinson’s Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Stereotaxic Surgery
2.2. Motor Behavioural Tests, Cohort 1
2.3. Tissue Processing
2.4. Immunohistochemistry, Cohort 1
2.5. Autoradiography, Cohort 1
2.6. In Situ Hybridization, Cohort 1
2.7. HPLC, Cohort 2
2.8. Statistics
3. Results
3.1. Transgene Expression
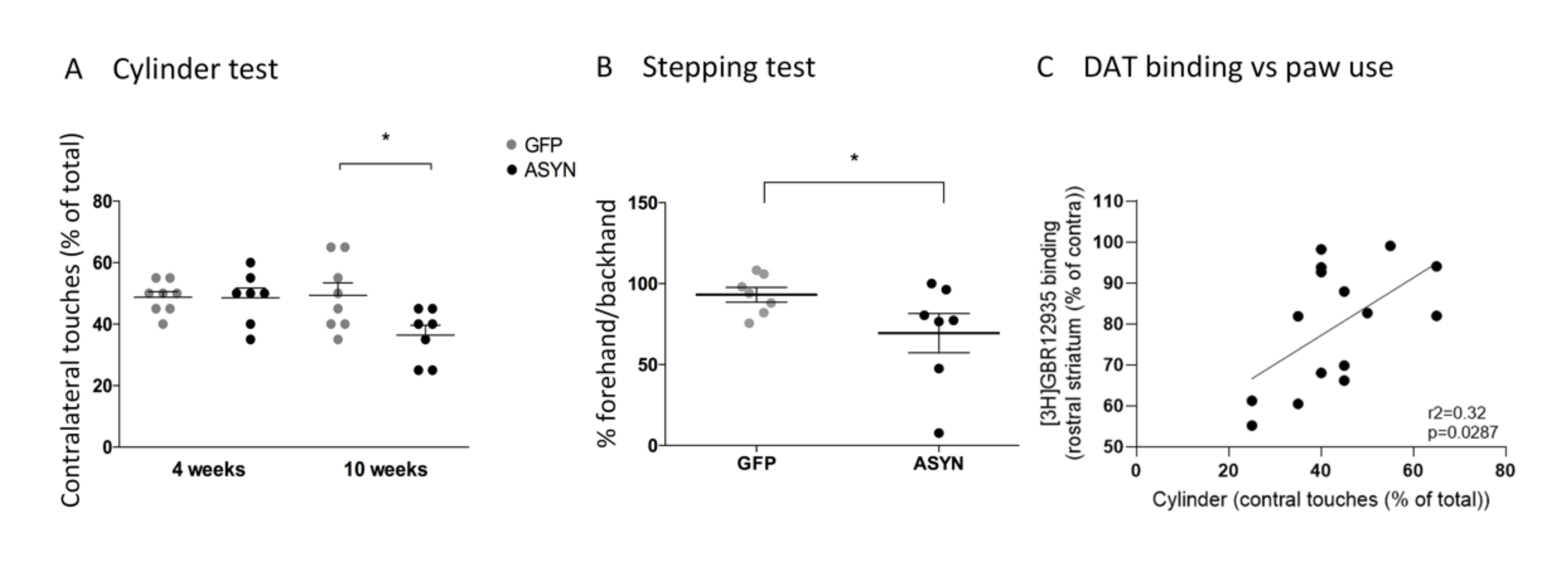
3.2. Behavioural Tests
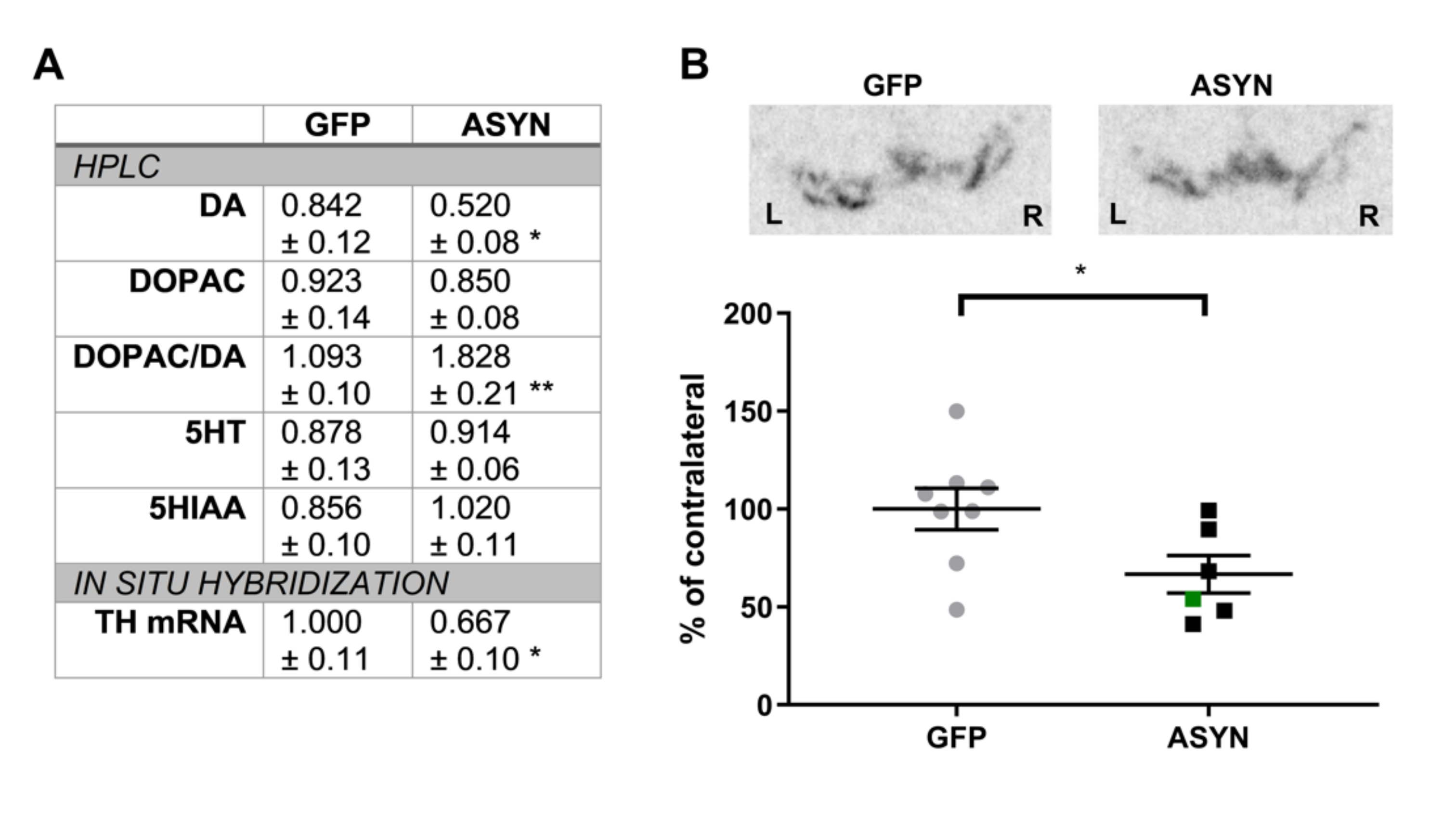
3.3. ASYN Expression Induced Reduction in the Nigral TH Expression and Striatal Dopamine Levels
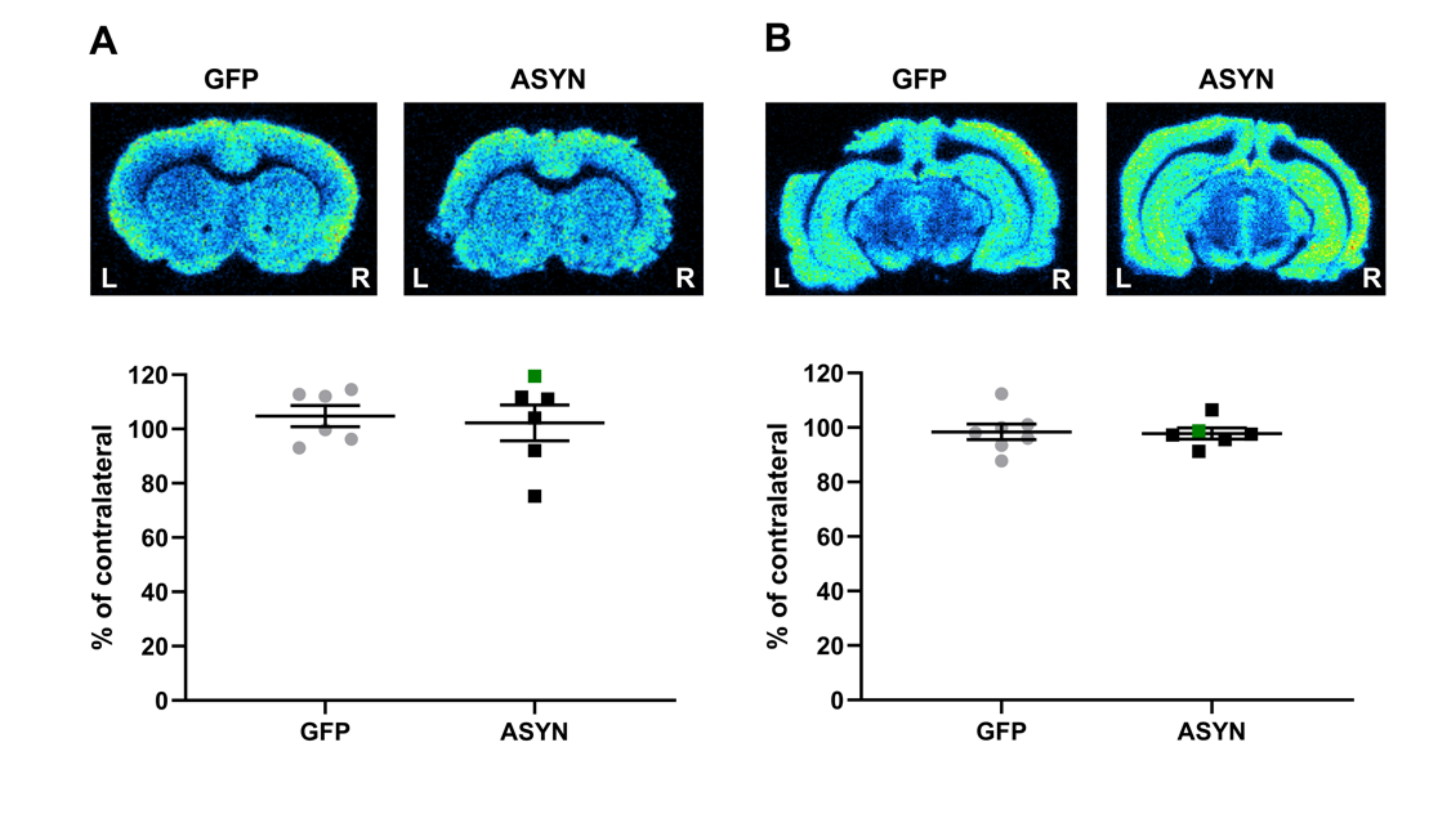
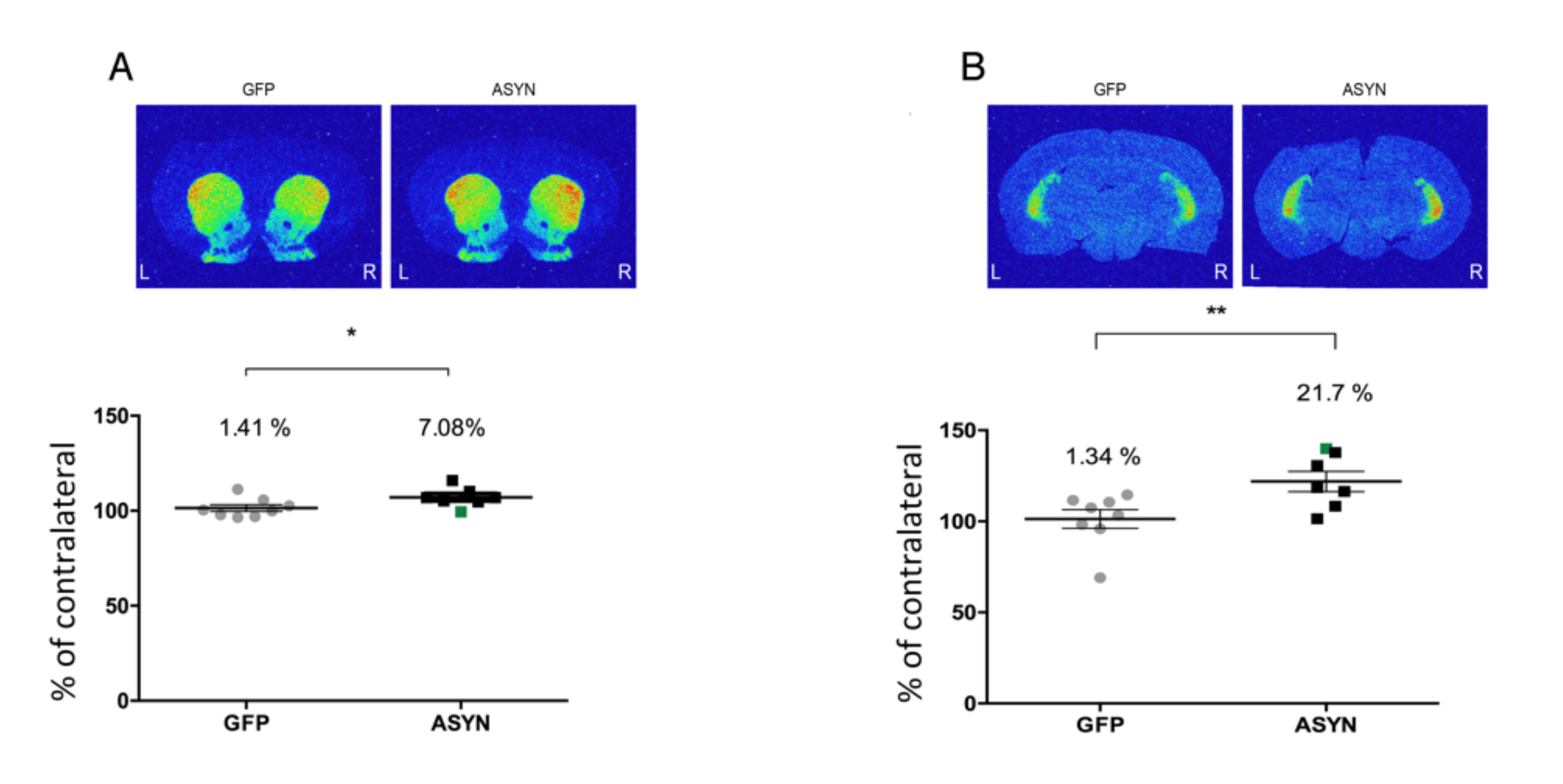
3.4. ASYN Overexpression in Nigrostriatal Neurons Induced Pre-and Post-Synaptic Changes in Striatum
3.5. ASYN Overexpression Induced Immune Activation in SN and Striatum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Bridi, J.C.; Hirth, F. Mechanisms of alpha-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Morrish, P.K.; Rakshi, J.S.; Bailey, D.L.; Sawle, G.V.; Brooks, D.J. Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with [18F]dopa PET. J. Neurol. Neurosurg. Psychiatry 1998, 64, 314–319. [Google Scholar] [CrossRef]
- Lee, C.S.; Samii, A.; Sossi, V.; Ruth, T.J.; Schulzer, M.; Holden, J.E.; Wudel, J.; Pal, P.K.; De la Fuente-Fernandez, R.; Calne, D.B.; et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson’s disease. Ann. Neurol. 2000, 47, 493–503. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, K.J.; Hsiao, I.T.; Hsieh, C.J.; Lin, W.Y.; Lu, C.S.; Wey, S.P.; Yen, T.C.; Kung, M.P.; Weng, Y.H. In vivo detection of monoaminergic degeneration in early Parkinson disease by (18)F-9-fluoropropyl-(+)-dihydrotetrabenzazine PET. J. Nucl. Med. 2014, 55, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Antonini, A.; Schwarz, J.; Oertel, W.H.; Pogarell, O.; Leenders, K.L. Long-term changes of striatal dopamine D2 receptors in patients with Parkinson’s disease: A study with positron emission tomography and [11C]raclopride. Mov. Disord. 1997, 12, 33–38. [Google Scholar] [CrossRef]
- Rinne, J.O.; Laihinen, A.; Ruottinen, H.; Ruotsalainen, U.; Nagren, K.; Lehikoinen, P.; Oikonen, V.; Rinne, U.K. Increased density of dopamine D2 receptors in the putamen, but not in the caudate nucleus in early Parkinson’s disease: A PET study with [11C]raclopride. J. Neurol. Sci. 1995, 132, 156–161. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: From pathology to imaging. Prog. Neurobiol. 2006, 80, 308–322. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Nutma, E.; Ceyzeriat, K.; Amor, S.; Tsartsalis, S.; Millet, P.; Owen, D.R.; Papadopoulos, V.; Tournier, B.B. Cellular sources of TSPO expression in healthy and diseased brain. Eur. J. Nucl. Med. Mol. Imaging 2021, 1–18. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin. Transl. Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Varnas, K.; Cselenyi, Z.; Jucaite, A.; Halldin, C.; Svenningsson, P.; Farde, L.; Varrone, A. PET imaging of [(11)C]PBR28 in Parkinson’s disease patients does not indicate increased binding to TSPO despite reduced dopamine transporter binding. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef]
- Ghadery, C.; Koshimori, Y.; Coakeley, S.; Harris, M.; Rusjan, P.; Kim, J.; Houle, S.; Strafella, A.P. Microglial activation in Parkinson’s disease using [(18)F]-FEPPA. J. Neuroinflammation 2017, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshimori, Y.; Ko, J.H.; Mizrahi, R.; Rusjan, P.; Mabrouk, R.; Jacobs, M.F.; Christopher, L.; Hamani, C.; Lang, A.E.; Wilson, A.A.; et al. Imaging Striatal Microglial Activation in Patients with Parkinson’s Disease. PLoS ONE 2015, 10, e0138721. [Google Scholar] [CrossRef]
- Phan, J.A.; Stokholm, K.; Zareba-Paslawska, J.; Jakobsen, S.; Vang, K.; Gjedde, A.; Landau, A.M.; Romero-Ramos, M. Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 6363. [Google Scholar] [CrossRef] [Green Version]
- Kirik, D.; Rosenblad, C.; Bjorklund, A.; Mandel, R.J. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: Intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J. Neurosci. 2000, 20, 4686–4700. [Google Scholar] [CrossRef]
- Olsson, M.; Nikkhah, G.; Bentlage, C.; Bjorklund, A. Forelimb akinesia in the rat Parkinson model: Differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J. Neurosci. 1995, 15, 3863–3875. [Google Scholar] [CrossRef]
- Minuzzi, L.; Olsen, A.K.; Bender, D.; Arnfred, S.; Grant, R.; Danielsen, E.H.; Cumming, P. Quantitative autoradiography of ligands for dopamine receptors and transporters in brain of Gottingen minipig: Comparison with results in vivo. Synapse 2006, 59, 211–219. [Google Scholar] [CrossRef]
- Christiansen, S.H.; Woldbye, D.P. Regulation of the galanin system by repeated electroconvulsive seizures in mice. J. Neurosci. Res. 2010, 88, 3635–3643. [Google Scholar] [CrossRef]
- Liebenberg, N.; Jensen, E.; Larsen, E.R.; Kousholt, B.S.; Pereira, V.S.; Fischer, C.W.; Wegener, G. A Preclinical Study of Casein Glycomacropeptide as a Dietary Intervention for Acute Mania. Int. J. Neuropsychopharmacol. 2018, 21, 473–484. [Google Scholar] [CrossRef]
- Chen, M.K.; Kuwabara, H.; Zhou, Y.; Adams, R.J.; Brasic, J.R.; McGlothan, J.L.; Verina, T.; Burton, N.C.; Alexander, M.; Kumar, A.; et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson’s disease. J. Neurochem. 2008, 105, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.; Fujinaga, M.; Kawamura, K.; Furutsuka, K.; Nengaki, N.; Shimoda, Y.; Shiomi, S.; Takei, M.; Hashimoto, H.; Yui, J.; et al. Dynamic Changes in Striatal mGluR1 But Not mGluR5 during Pathological Progression of Parkinson’s Disease in Human Alpha-Synuclein A53T Transgenic Rats: A Multi-PET Imaging Study. J. Neurosci. 2016, 36, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Perren, A.; Toelen, J.; Casteels, C.; Macchi, F.; Van Rompuy, A.S.; Sarre, S.; Casadei, N.; Nuber, S.; Himmelreich, U.; Osorio Garcia, M.I.; et al. Longitudinal follow-up and characterization of a robust rat model for Parkinson’s disease based on overexpression of alpha-synuclein with adeno-associated viral vectors. Neurobiol. Aging 2015, 36, 1543–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Kouranova, E.; Cui, X.; Mach, R.H.; Xu, J. Regulation of dopamine presynaptic markers and receptors in the striatum of DJ-1 and Pink1 knockout rats. Neurosci. Lett. 2013, 557, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE 2010, 5, e9956. [Google Scholar]
- Cai, Z.; Li, S.; Matuskey, D.; Nabulsi, N.; Huang, Y. PET imaging of synaptic density: A new tool for investigation of neuropsychiatric diseases. Neurosci. Lett. 2019, 691, 44–50. [Google Scholar] [CrossRef]
- Delva, A.; Van Weehaeghe, D.; Koole, M.; Van Laere, K.; Vandenberghe, W. Loss of Presynaptic Terminal Integrity in the Substantia Nigra in Early Parkinson’s Disease. Mov. Disord. 2020, 35, 1977–1986. [Google Scholar] [CrossRef]
- Matuskey, D.; Tinaz, S.; Wilcox, K.C.; Naganawa, M.; Toyonaga, T.; Dias, M.; Henry, S.; Pittman, B.; Ropchan, J.; Nabulsi, N.; et al. Synaptic Changes in Parkinson Disease Assessed with in vivo Imaging. Ann. Neurol. 2020, 87, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.; Pagano, G.; De Natale, E.R.; Mansur, A.; Caminiti, S.P.; Polychronis, S.; Middleton, L.T.; Price, G.; Schmidt, K.F.; Gunn, R.N.; et al. Mitochondrial Complex 1, Sigma 1, and Synaptic Vesicle 2A in Early Drug-Naive Parkinson’s Disease. Mov. Disord. 2020, 35, 1416–1427. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Jacobsen, J.; Lillethorup, T.P.; Schacht, A.C.; Simonsen, M.; Romero-Ramos, M.; Brooks, D.J.; Landau, A.M. In vivo imaging of synaptic SV2A protein density in healthy and striatal-lesioned rats with [11C]UCB-J PET. J. Cereb. Blood Flow Metab. 2021, 41, 819–830. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Ferreira, S.A.; Schacht, A.C.; Jacobsen, J.; Simonsen, M.; Betzer, C.; Jensen, P.H.; Brooks, D.J.; Landau, A.M.; Romero-Ramos, M. PET imaging reveals early and progressive dopaminergic deficits after intra-striatal injection of preformed alpha-synuclein fibrils in rats. Neurobiol. Dis. 2020, 149, 105229. [Google Scholar] [CrossRef]
- Groves, P.M.; Linder, J.C.; Young, S.J. 5-hydroxydopamine-labeled dopaminergic axons: Three-dimensional reconstructions of axons, synapses and postsynaptic targets in rat neostriatum. Neuroscience 1994, 58, 593–604. [Google Scholar] [CrossRef]
- Uchigashima, M.; Ohtsuka, T.; Kobayashi, K.; Watanabe, M. Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc. Natl. Acad. Sci. USA 2016, 113, 4206–4211. [Google Scholar] [CrossRef] [Green Version]
- Molinet-Dronda, F.; Gago, B.; Quiroga-Varela, A.; Juri, C.; Collantes, M.; Delgado, M.; Prieto, E.; Ecay, M.; Iglesias, E.; Marin, C.; et al. Monoaminergic PET imaging and histopathological correlation in unilateral and bilateral 6-hydroxydopamine lesioned rat models of Parkinson’s disease: A longitudinal in-vivo study. Neurobiol. Dis. 2015, 77, 165–172. [Google Scholar] [CrossRef]
- Du, G.; Lewis, M.M.; Sen, S.; Wang, J.; Shaffer, M.L.; Styner, M.; Yang, Q.X.; Huang, X. Imaging nigral pathology and clinical progression in Parkinson’s disease. Mov. Disord. 2012, 27, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Kirik, D.; Rosenblad, C.; Bjorklund, A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp. Neurol. 1998, 152, 259–277. [Google Scholar] [CrossRef]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Bjorklund, A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef] [Green Version]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflammation 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Barkholt, P.; Sanchez-Guajardo, V.; Kirik, D.; Romero-Ramos, M. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience 2012, 208, 85–96. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Rodriguez-Chinchilla, T.; Quiroga-Varela, A.; Molinet-Dronda, F.; Belloso-Iguerategui, A.; Merino-Galan, L.; Jimenez-Urbieta, H.; Gago, B.; Rodriguez-Oroz, M.C. [(18)F]-DPA-714 PET as a specific in vivo marker of early microglial activation in a rat model of progressive dopaminergic degeneration. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2602–2612. [Google Scholar] [CrossRef]
- Crabbe, M.; Van der Perren, A.; Kounelis, S.; Lavreys, T.; Bormans, G.; Baekelandt, V.; Casteels, C.; Van Laere, K. Temporal changes in neuroinflammation and brain glucose metabolism in a rat model of viral vector-induced alpha-synucleinopathy. Exp. Neurol. 2019, 320, 112964. [Google Scholar] [CrossRef]
- Maia, S.; Arlicot, N.; Vierron, E.; Bodard, S.; Vergote, J.; Guilloteau, D.; Chalon, S. Longitudinal and parallel monitoring of neuroinflammation and neurodegeneration in a 6-hydroxydopamine rat model of Parkinson’s disease. Synapse 2012, 66, 573–583. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Stokholm, M.G.; Iranzo, A.; Ostergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Lillethorup, T.P.; Glud, A.N.; Landeck, N.; Alstrup, A.K.O.; Jakobsen, S.; Vang, K.; Doudet, D.J.; Brooks, D.J.; Kirik, D.; Hinz, R.; et al. In vivo quantification of glial activation in minipigs overexpressing human alpha-synuclein. Synapse 2018, 72, e22060. [Google Scholar] [CrossRef] [Green Version]
- Francois, C.; Percheron, G.; Yelnik, J. Localization of nigrostriatal, nigrothalamic and nigrotectal neurons in ventricular coordinates in macaques. Neuroscience 1984, 13, 61–76. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokholm, K.; Thomsen, M.B.; Phan, J.-A.; Møller, L.K.; Bay-Richter, C.; Christiansen, S.H.; Woldbye, D.P.D.; Romero-Ramos, M.; Landau, A.M. α-Synuclein Overexpression Increases Dopamine D2/3 Receptor Binding and Immune Activation in a Model of Early Parkinson’s Disease. Biomedicines 2021, 9, 1876. https://doi.org/10.3390/biomedicines9121876
Stokholm K, Thomsen MB, Phan J-A, Møller LK, Bay-Richter C, Christiansen SH, Woldbye DPD, Romero-Ramos M, Landau AM. α-Synuclein Overexpression Increases Dopamine D2/3 Receptor Binding and Immune Activation in a Model of Early Parkinson’s Disease. Biomedicines. 2021; 9(12):1876. https://doi.org/10.3390/biomedicines9121876
Chicago/Turabian StyleStokholm, Kathrine, Majken Borup Thomsen, Jenny-Ann Phan, Line K. Møller, Cecilie Bay-Richter, Søren H. Christiansen, David P. D. Woldbye, Marina Romero-Ramos, and Anne M. Landau. 2021. "α-Synuclein Overexpression Increases Dopamine D2/3 Receptor Binding and Immune Activation in a Model of Early Parkinson’s Disease" Biomedicines 9, no. 12: 1876. https://doi.org/10.3390/biomedicines9121876
APA StyleStokholm, K., Thomsen, M. B., Phan, J.-A., Møller, L. K., Bay-Richter, C., Christiansen, S. H., Woldbye, D. P. D., Romero-Ramos, M., & Landau, A. M. (2021). α-Synuclein Overexpression Increases Dopamine D2/3 Receptor Binding and Immune Activation in a Model of Early Parkinson’s Disease. Biomedicines, 9(12), 1876. https://doi.org/10.3390/biomedicines9121876