
Non-Genetically Encoded Epitopes Are Relevant Targets in Autoimmune Diabetes



Abstract
:1. Introduction
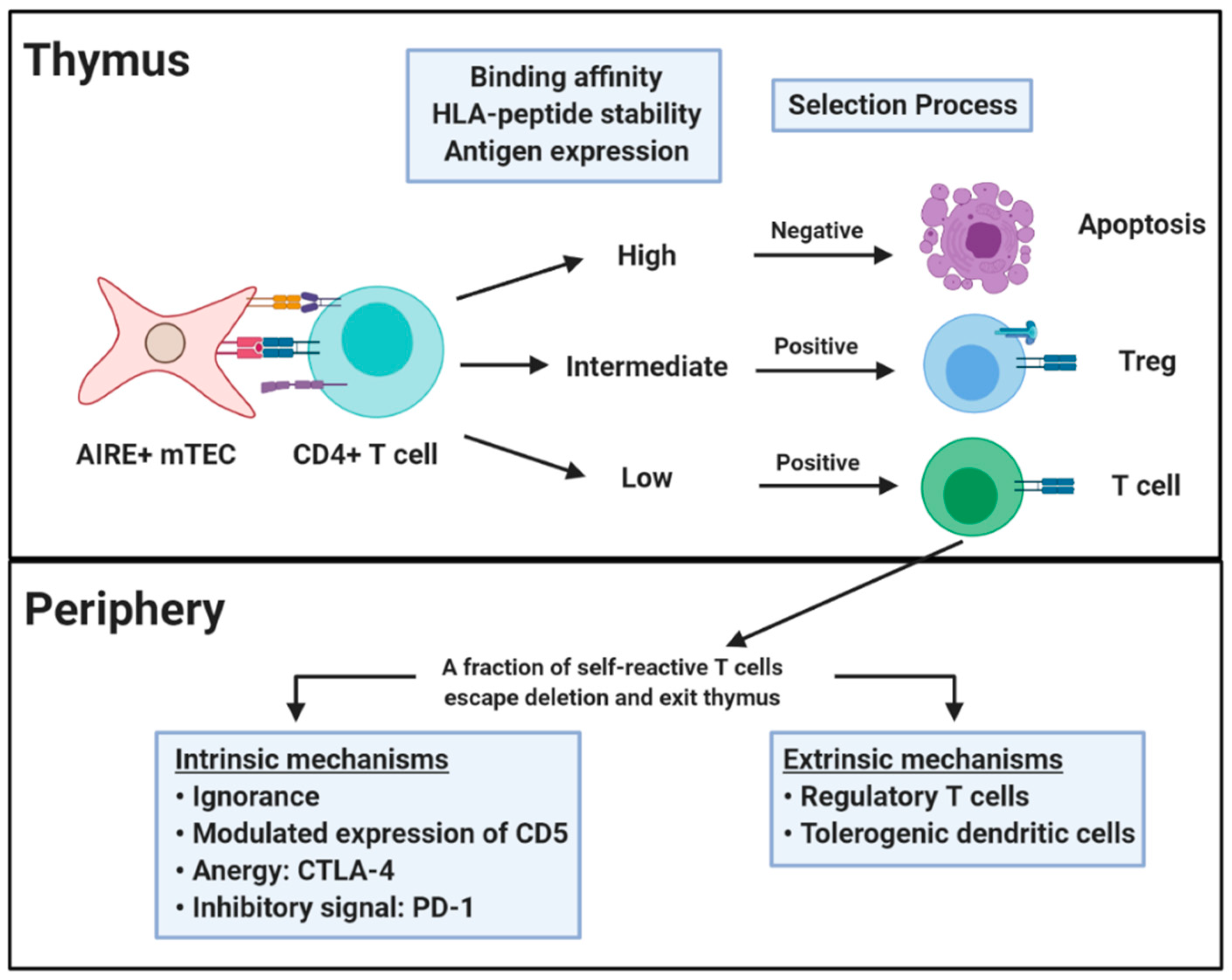
2. Central and Peripheral Tolerance
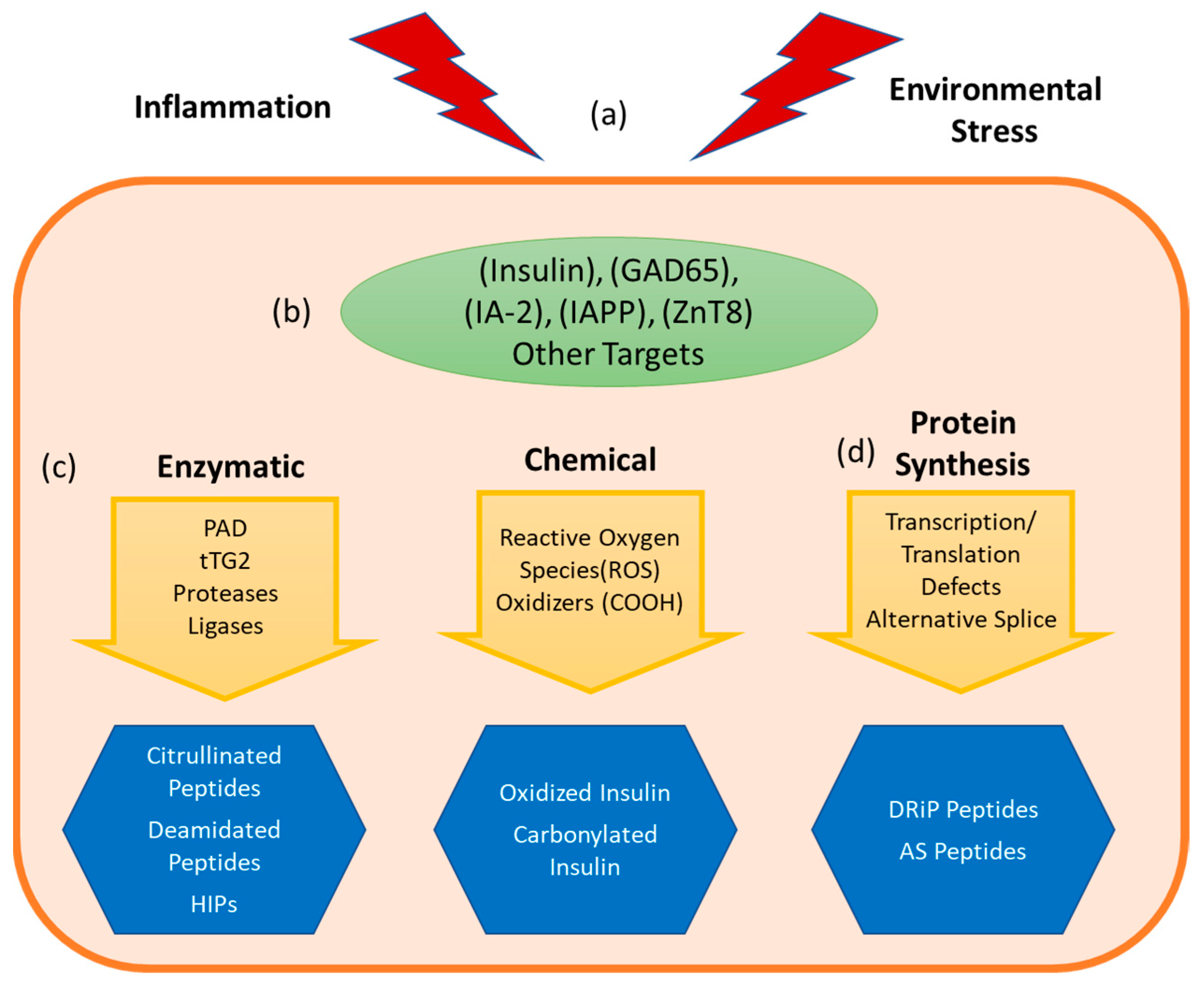
3. Non-Genetically Encoded Neo-Antigens and Epitopes
3.1. Insulin Derived Neo-Antigens
3.1.1. Insulin Oxidation
3.1.2. Hybrid Insulin Peptides
3.1.3. DRiP
3.2. Enzymatically Generated Neo-Antigens
3.3. Neo-antigens Generated through Alternative Splicing
4. Inducing Antigen Specific Tolerance Using Peptide Neo-Epitopes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkinson, M.A. The Pathogenesis and Natural History of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, M.; Towns, R.; Eisenbarth, G.S. Humoral Autoimmunity in Type 1 Diabetes: Prediction, Significance, and Detection of Distinct Disease Subtypes. Cold Spring Harb. Perspect. Med. 2012, 2, a012831. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Rewers, M.; Gianani, R.; Kawasaki, E.; Zhang, Y.; Verge, C.; Chase, P.; Klingensmith, G.; Erlich, H.; Norris, J.; et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J. Clin. Endocrinol. Metab. 1996, 81, 4264–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, E.A.M.R.; Kent, S.C.; DiLorenzo, T.P. T cell epitopes and neo-epitopes in type 1 diabetes: A comprehensive update and re-appraisal. Diabetes 2020, 69, 1311–1335. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.J. Cellular immunity to human insulin in individuals at high risk for the development of Type I diabetes mellitus. J. Autoimmun. 1990, 3, 321–327. [Google Scholar] [CrossRef]
- Alleva, D.G.; Crowe, P.D.; Jin, L.; Kwok, W.W.; Ling, N.; Gottschalk, M.; Conlon, P.J.; Gottlieb, P.A.; Putnam, A.L.; Gaur, A. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J. Clin. Investig. 2001, 107, 173–180. [Google Scholar] [CrossRef]
- Nepom, G.T.; Lippolis, J.D.; White, F.M.; Masewicz, S.; Marto, J.A.; Herman, A.; Luckey, C.J.; Falk, B.; Shabanowitz, J.; Hunt, D.F.; et al. Identification and modulation of a naturally processed T cell epitope from the diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65). Proc. Natl. Acad. Sci. USA 2001, 98, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, K.A.; Gulati, K.; Richardson, C.C.; Morgan, D.; Bodansky, H.J.; Feltbower, R.G.; Christie, M.R. HLA-DR4-associated T and B cell responses to specific determinants on the IA-2 autoantigen in type 1 diabetes. J. Immunol. 2014, 193, 4448–4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; Von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Babon, J.A.B.; DeNicola, M.E.; Blodgett, D.M.; Crèvecoeur, I.; Buttrick, T.S.; Maehr, R.; Bottino, R.; Naji, A.; Kaddis, J.; Elyaman, W.; et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 2016, 22, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Erlich, H.; Valdes, A.M.; Noble, J.; Carlson, J.A.; Varney, M.; Concannon, P.; Mychaleckyj, J.C.; Todd, J.A.; Bonella, P.; Fear, A.L.; et al. HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Diabetes 2008, 57, 1084–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [Green Version]
- Price, P.; Witt, C.; Allock, R.; Sayer, D.; Garlepp, M.; Kok, C.C.; French, M.; Mallal, S.; Christiansen, F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 1999, 167, 257–274. [Google Scholar] [CrossRef]
- Moustakas, A.K.; Papadopoulos, G.K. Use of MHC II structural features in the design of vaccines for organ-specific autoimmune diseases. Curr. Pharm. Des. 2009, 15, 3262–3273. [Google Scholar] [CrossRef]
- Sharon, E.; Sibener, L.V.; Battle, A.; Fraser, H.B.; Garcia, L.V.S.K.C.; Pritchard, E.S.H.B.F.J.K. Genetic variation in MHC proteins is associated with T cell receptor expression biases. Nat. Genet. 2016, 48, 995–1002. [Google Scholar] [CrossRef]
- Harbige, J.; Eichmann, M.; Peakman, M. New insights into non-conventional epitopes as T cell targets: The missing link for breaking immune tolerance in autoimmune disease? J. Autoimmun. 2017, 84, 12–20. [Google Scholar] [CrossRef]
- James, E.A.; Pietropaolo, M.; Mamula, M.J. Immune Recognition of β-Cells: Neoepitopes as Key Players in the Loss of Tolerance. Diabetes 2018, 67, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Doyle, H.A.; Mamula, M.J. Autoantigenesis: The evolution of protein modifications in autoimmune disease. Curr. Opin. Immunol. 2012, 24, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Ciofani, M.; Zúñiga-Pflücker, J.C. The Thymus as an Inductive Site for T Lymphopoiesis. Annu. Rev. Cell Dev. Biol. 2007, 23, 463–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodnow, C.C.; Sprent, J.; Groth, B.F.D.S.; Vinuesa, C.G. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nat. Cell Biol. 2005, 435, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, A.M.; Bevan, M.J. Central tolerance: Good but imperfect. Immunol. Rev. 2006, 209, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, A.M.; Bevan, M.J. Central Tolerance to Tissue-specific Antigens Mediated by Direct and Indirect Antigen Presentation. J. Exp. Med. 2004, 200, 1039–1049. [Google Scholar] [CrossRef] [Green Version]
- Derbinski, J.; Schulte, A.; Kyewski, B.; Klein, L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2001, 2, 1032–1039. [Google Scholar] [CrossRef]
- Nemazee, D.; Hogquist, K.A. Antigen receptor selection by editing or downregulation of V(D)J recombination. Curr. Opin. Immunol. 2003, 15, 182–189. [Google Scholar] [CrossRef]
- Danke, N.A.; Koelle, D.M.; Yee, C.; Beheray, S.; Kwok, W.W. Autoreactive T cells in healthy individuals. J. Immunol. 2004, 172, 5967–5972. [Google Scholar] [CrossRef] [Green Version]
- Busch, R.; De Riva, A.; Hadjinicolaou, A.V.; Jiang, W.; Hou, T.; Mellins, E.D. On the perils of poor editing: Regulation of peptide loading by HLA-DQ and H2-A molecules associated with celiac disease and type 1 diabetes. Expert Rev. Mol. Med. 2012, 14, e15. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, R.A.; Liu, A.W.; Nepom, G.T.; Kwok, W.W. Exceptional stability of the HLA-DQA1*0102/DQB1*0602 alpha beta protein dimer, the class II MHC molecule associated with protection from insulin-dependent diabetes mellitus. J. Immunol. 1998, 161, 6439–6445. [Google Scholar]
- Zhang, M.; Vacchio, M.S.; Vistica, B.P.; Lesage, S.; Egwuagu, C.E.; Yu, C.-R.; Gelderman, M.P.; Kennedy, M.C.; Wawrousek, E.F.; Gery, I. T cell tolerance to a neo-self antigen expressed by thymic epithelial cells: The soluble form is more effective than the membrane-bound form. J. Immunol. 2003, 170, 3954–3962. [Google Scholar] [CrossRef] [Green Version]
- Vafiadis, P.; Bennett, S.T.; Todd, J.A.; Nadeau, J.; Grabs, R.; Goodyer, C.G.; Wickramasinghe, S.; Colle, E.; Polychronakos, C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 1997, 15, 289–292. [Google Scholar] [CrossRef]
- Durinovic-Belló, I.; Wu, R.P.; Gersuk, V.H.; Sanda, S.; Shilling, H.G.; Nepom, G.T. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun. 2010, 11, 188–193. [Google Scholar] [CrossRef]
- Walker, L.S.; Abbas, A.K. The enemy within: Keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2002, 2, 11–19. [Google Scholar] [CrossRef]
- Wong, P.; Barton, G.M.; Forbush, K.A.; Rudensky, A.Y. Dynamic Tuning of T Cell Reactivity by Self-Peptide–Major Histocompatibility Complex Ligands. J. Exp. Med. 2001, 193, 1179–1188. [Google Scholar] [CrossRef]
- Smith, K.; Seddon, B.; Purbhoo, M.A.; Zamoyska, R.; Fisher, A.G.; Merkenschlager, M. Sensory Adaptation in Naive Peripheral CD4 T Cells. J. Exp. Med. 2001, 194, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.J.I.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The Programmed Death-1 (PD-1) Pathway Regulates Autoimmune Diabetes in Nonobese Diabetic (NOD) Mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.K. Regulatory T cells overturned: The effectors fight back. Immunology 2009, 126, 466–474. [Google Scholar] [CrossRef]
- Marre, M.L.; McGinty, J.W.; Chow, I.-T.; DeNicola, M.E.; Beck, N.W.; Kent, S.C.; Powers, A.C.; Bottino, R.; Harlan, D.M.; Greenbaum, C.J.; et al. Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4+T Cells in Patients With Type 1 Diabetes. Diabetes 2018, 67, 1356–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, H.A.; Mamula, M.J. Posttranslational Modifications of Self-Antigens. Ann. N. Y. Acad. Sci. 2005, 1050, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.; Steiner, D. The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. [Google Scholar] [CrossRef]
- Van Lommel, L.; Janssens, K.; Quintens, R.; Tsukamoto, K.; Mierde, D.V.; Lemaire, K.; Denef, C.; Jonas, J.-C.; Martens, G.; Pipeleers, D.; et al. Probe-Independent and Direct Quantification of Insulin mRNA and Growth Hormone mRNA in Enriched Cell Preparations. Diabetes 2006, 55, 3214–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serreze, D.V.; Wasserfall, C.; Ottendorfer, E.W.; Stalvey, M.; Pierce, M.A.; Gauntt, C.; O’Donnell, B.; Flanagan, J.B.; Campbell-Thompson, M.; Ellis, T.M.; et al. Diabetes Acceleration or Prevention by a Coxsackievirus B4 Infection: Critical Requirements for both Interleukin-4 and Gamma Interferon. J. Virol. 2005, 79, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serreze, D.V.; Ottendorfer, E.W.; Ellis, T.M.; Gauntt, C.J.; Atkinson, M.A. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000, 49, 708–711. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; De Beeck, A.O.; De Brachène, A.C.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Barthson, J.; Marhfour, I.; Ortis, F.; Naamane, N.; Igoillo-Esteve, M.; Gysemans, C.; Mathieu, C.; Kitajima, S.; Marchetti, P.; et al. Pancreatic β-cells activate a JunB/ATF3-dependent survival pathway during inflammation. Oncogene 2011, 31, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Marré, M.L.; Profozich, J.L.; Coneybeer, J.T.; Geng, X.; Bertera, S.; Ford, M.J.; Trucco, M.; Piganelli, J.D. Inherent ER stress in pancreatic islet β cells causes self-recognition by autoreactive T cells in type 1 diabetes. J. Autoimmun. 2016, 72, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Rondas, D.; Crèvecoeur, I.; D’Hertog, W.; Ferreira, G.B.; Staes, A.; Garg, A.D.; Eizirik, D.L.; Agostinis, P.; Gevaert, K.; Overbergh, L.; et al. Citrullinated Glucose-Regulated Protein 78 Is an Autoantigen in Type 1 Diabetes. Diabetes 2014, 64, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Duque, S.; Azoury, M.E.; Colli, M.L.; Afonso, G.; Turatsinze, J.-V.; Nigi, L.; Lalanne, A.I.; Sebastiani, G.; Carré, A.; Pinto, S.; et al. Conventional and Neo-antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab. 2018, 28, 946–960.e6. [Google Scholar] [CrossRef] [Green Version]
- Kracht, M.J.L.; Van Lummel, M.; Nikolic, T.; Joosten, A.M.; Laban, S.; Van Der Slik, A.R.; Van Veelen, P.A.; Carlotti, F.; De Koning, F.C.E.J.P.; Hoeben, R.C.; et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat. Med. 2017, 23, 501–507. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cnop, M. ER Stress in Pancreatic Cells: The Thin Red Line Between Adaptation and Failure. Sci. Signal. 2010, 3, pe7. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, R.E.; Winston, B.; Cohen, G.; Barden, H. Alloxan-induced diabetes—evidence for hydroxyl radical as a cytotoxic intermediate. Biochem. Pharmacol. 1976, 25, 1085–1092. [Google Scholar] [CrossRef]
- Takasu, N.; Komiya, I.; Asawa, T.; Nagasawa, Y.; Yamada, T. Streptozocin- and Alloxan-Induced H2O2 Generation and DNA Fragmentation in Pancreatic Islets: H2O2 as Mediator for DNA Fragmentation. Diabetes 1991, 40, 1141–1145. [Google Scholar] [CrossRef]
- Yu, L.; Dong, F.; Miao, D.; Fouts, A.R.; Wenzlau, J.M.; Steck, A.K. Proinsulin/Insulin Autoantibodies Measured with Electrochemiluminescent Assay Are the Earliest Indicator of Prediabetic Islet Autoimmunity. Diabetes Care 2013, 36, 2266–2270. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Abiru, N.; Moriyama, H.; Babaya, N.; Liu, E.; Miao, D.; Yu, L.; Wegmann, D.R.; Hutton, J.C.; Elliott, J.F.; et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nat. Cell Biol. 2005, 435, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Blahnik, G.; Uchtenhagen, H.; Chow, I.-T.; Speake, C.; Greenbaum, C.; Kwok, W.W.; James, E.A. Analysis of pancreatic beta cell specific CD4+ T cells reveals a predominance of proinsulin specific cells. Cell. Immunol. 2019, 335, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Mannering, S.I.; Harrison, L.C.; Williamson, N.A.; Morris, J.S.; Thearle, D.J.; Jensen, K.P.; Kay, T.W.; Rossjohn, J.; Falk, B.A.; Nepom, G.T.; et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J. Exp. Med. 2005, 202, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Strollo, R.; Vinci, C.; Arshad, M.H.; Perrett, D.; Tiberti, C.; Chiarelli, F.; Napoli, N.; Pozzilli, P.; Nissim, A. Antibodies to post-translationally modified insulin in type 1 diabetes. Diabetologia 2015, 58, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strollo, R.; Vinci, C.; Napoli, N.; Pozzilli, P.; Ludvigsson, J.; Nissim, A. Antibodies to post-translationally modified insulin as a novel biomarker for prediction of type 1 diabetes in children. Diabetologia 2017, 60, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Strollo, R.; Vinci, C.; Napoli, N.; Fioriti, E.; Maddaloni, E.; Åkerman, L.; Casas, R.; Pozzilli, P.; Ludvigsson, J.; Nissim, A. Antibodies to oxidized insulin improve prediction of type 1 diabetes in children with positive standard islet autoantibodies. Diabetes Metab. Res. Rev. 2019, 35, e3132. [Google Scholar] [CrossRef]
- Hecker, M.; Wagner, A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2017, 41, 29–38. [Google Scholar] [CrossRef]
- Domínguez, C.; Ruiz, E.; Gussinye, M.; Carrascosa, A. Oxidative Stress at Onset and in Early Stages of Type 1 Diabetes in Children and Adolescents. Diabetes Care 1998, 21, 1736–1742. [Google Scholar] [CrossRef]
- Sanchez-Quesada, J.L. Modified low-density lipoproteins as biomarkers in diabetes and metabolic syndrome. Front. Biosci. 2018, 23, 1220–1240. [Google Scholar] [CrossRef] [Green Version]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.L.; Jamison, B.L.; Haskins, K. Hybrid insulin peptides are neo-epitopes for CD4 T cells in autoimmune diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 195–200. [Google Scholar] [CrossRef]
- Arribas-Layton, D.; Guyer, P.; Delong, T.; Dang, M.; Chow, I.-T.; Speake, C.; Greenbaum, C.J.; Kwok, W.W.; Baker, R.L.; Haskins, K.; et al. Hybrid Insulin Peptides Are Recognized by Human T Cells in the Context of DRB1*04:01. Diabetes 2020, 69, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Kishida, K.; Matsumoto, M.; Matsuoka, S.; Kohyama, M.; Suenaga, T.; Arase, H. A TCR-like antibody against a proinsulin-containing fusion peptide ameliorates type 1 diabetes in NOD mice. Biochem. Biophys. Res. Commun. 2021, 534, 680–686. [Google Scholar] [CrossRef]
- Laban, S.; Suwandi, J.S.; Van Unen, V.; Pool, J.; Wesselius, J.; Höllt, T.; Pezzotti, N.; Vilanova, A.; Lelieveldt, B.P.F.; Roep, B.O. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PLoS ONE 2018, 13, e0200818. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; James, E.A. Immune recognition of citrullinated epitopes. Immunology 2016, 149, 131–138. [Google Scholar] [CrossRef]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction with the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J. Immunol. 2003, 171, 538–541. [Google Scholar] [CrossRef] [Green Version]
- James, E.A.; Rieck, M.; Pieper, J.; Gebe, J.A.; Yue, B.B.; Tatum, M.; Peda, M.; Sandin, C.; Klareskog, L.; Malmström, V.; et al. Citrulline-Specific Th1 Cells Are Increased in Rheumatoid Arthritis and Their Frequency Is Influenced by Disease Duration and Therapy. Arthritis Rheumatol. 2014, 66, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- James, E.A.; Moustakas, A.K.; Bui, J.; Papadopoulos, G.K.; Bondinas, G.P.; Buckner, J.H.; Kwok, W.W. HLA-DR1001 presents “altered-self” peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets. Arthritis Rheum. 2010, 62, 2909–2918. [Google Scholar] [CrossRef] [Green Version]
- Van Venrooij, W.J.; Pruijn, G.J.M. Citrullination: A small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000, 2, 249–251. [Google Scholar] [CrossRef] [Green Version]
- McGinty, J.W.; Chow, I.-T.; Greenbaum, C.; Odegard, J.; Kwok, W.W.; James, E.A. Recognition of Posttranslationally Modified GAD65 Epitopes in Subjects With Type 1 Diabetes. Diabetes 2014, 63, 3033–3040. [Google Scholar] [CrossRef] [Green Version]
- Buitinga, M.; Callebaut, A.; Sodré, F.M.C.; Crèvecoeur, I.; Blahnik-Fagan, G.; Yang, M.-L.; Bugliani, M.; Arribas-Layton, D.; Marré, M.; Cook, D.P.; et al. Inflammation-Induced Citrullinated Glucose-Regulated Protein 78 Elicits Immune Responses in Human Type 1 Diabetes. Diabetes 2018, 67, 2337–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Wucherpfennig, K.W.; Wiley, D.C. Structure of a human insulin peptide–HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat. Immunol. 2001, 2, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Godkin, A.; Friede, T.; Davenport, M.; Stevanovic, S.; Willis, A.; Jewell, D.; Hill, A.; Rammensee, H.G. Use of eluted peptide sequence data to identify the binding characteristics of peptides to the insulin-dependent diabetes susceptibility allele HLA-DQ8 (DQ 3.2). Int. Immunol. 1997, 9, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.P.; Papadopoulos, G.K.; Kwok, W.W.; Moustakas, A.K.; Bondinas, G.P.; Larsson, H.E.; Ludvigsson, J.; Marcus, C.; Samuelsson, U.; Wang, R.; et al. Motifs of Three HLA-DQ Amino Acid Residues (α44, β57, β135) Capture Full Association with the Risk of Type 1 Diabetes in DQ2 and DQ8 Children. Diabetes 2020, 69, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Van Lummel, M.; Duinkerken, G.; Van Veelen, P.A.; De Ru, A.; Cordfunke, R.; Zaldumbide, A.; Gómez-Touriño, I.; Arif, S.; Peakman, M.; Drijfhout, J.W.; et al. Posttranslational Modification of HLA-DQ Binding Islet Autoantigens in Type 1 Diabetes. Diabetes 2013, 63, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Sollid, L.M.; Tye-Din, J.A.; Qiao, S.-W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Update 2020: Nomenclature and listing of celiac disease–relevant gluten epitopes recognized by CD4+ T cells. Immunogenetics 2019, 72, 85–88. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The Human Pancreatic Islet Transcriptome: Expression of Candidate Genes for Type 1 Diabetes and the Impact of Pro-Inflammatory Cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Colli, M.L.; Ramos-Rodríguez, M.; Nakayasu, E.S.; Alvelos, M.I.; Lopes, M.; Hill, J.L.; Turatsinze, J.V.; de Brachène, A.C.; Russell, M.A.; Raurell-Vila, H.; et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dogra, R.S.; Vaidyanathan, P.; Prabakar, K.R.; Marshall, K.E.; Hutton, J.C.; Pugliese, A. Alternative splicing of G6PC2, the gene coding for the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), results in differential expression in human thymus and spleen compared with pancreas. Diabetologia 2006, 49, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Liu, Y.-F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, C.; Liu, G.Y.; Anderton, S.M.; Metzler, B.; Wraith, D.C. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: A role for IL-10. Int. Immunol. 1999, 11, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nat. Cell Biol. 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Pearson, R.M.; Casey, L.M.; Hughes, K.R.; Wang, L.Z.; North, M.G.; Getts, D.R.; Miller, S.D.; Shea, L.D. Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles Using Peptide-Polymer Bioconjugates. Mol. Ther. 2017, 25, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Saito, E.; Gurczynski, S.J.; Kramer, K.R.; Wilke, C.A.; Miller, S.D.; Moore, B.B.; Shea, L.D. Modulating lung immune cells by pulmonary delivery of antigen-specific nanoparticles to treat autoimmune disease. Sci. Adv. 2020, 6, eabc9317. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.; Santamaria, P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018, 48, 751–756. [Google Scholar] [CrossRef]
- Ruiz, P.J.; Garren, H.; Ruiz, I.U.; Hirschberg, D.L.; Nguyen, L.V.; Karpuj, M.V.; Cooper, M.T.; Mitchell, D.J.; Fathman, C.G.; Steinman, L. Suppressive immunization with DNA encoding a self-peptide prevents autoimmune disease: Modulation of T cell costimula-tion. J. Immunol. 1999, 162, 3336. [Google Scholar]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef]
- Thomas, R. Dendritic cells and the promise of antigen-specific therapy in rheumatoid arthritis. Arthritis Res. 2013, 15, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benham, H.; Nel, H.J.; Law, S.C.; Mehdi, A.M.; Street, S.; Ramnoruth, N.; Pahau, H.; Lee, B.T.; Ng, J.; Brunck, M.E.G.; et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype–positive rheumatoid arthritis patients. Sci. Transl. Med. 2015, 7, 290ra87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firdessa-Fite, R.; Johnson, S.N.; Leon, M.A.; Khosravi-Maharlooei, M.; Baker, R.L.; Sestak, J.O.; Berkland, C.; Creusot, R.J. Soluble Antigen Arrays Efficiently Deliver Peptides and Arrest Spontaneous Autoimmune Diabetes. Diabetes 2021, db200845. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Technique | Administration Route 3 | Effect | References |
|---|---|---|---|
| Direct peptide delivery | INH/NAS/ID | IL-10 secretion by ag-specific CD4+ T cells | [88,89] |
| p/MHC 1 | IV | Induction of ag-specific Tr1-like 4 cells and regulatory network | [90] |
| p/MHC nanoparticle 1 | INH/IT | Induction of tolerogenic APCs and reduced autoreactive T cell proliferation | [91,92] |
| DNA encoding self-peptide | IM | Reduced IFN-γ and altered co-stimulation. | [94] |
| Tolerogenic DCs 2 | PO/ID | Improved regulatory/effector T cell ratio | [96,97] |
| Soluble antigen arrays | SC | Induction of IL-10 and exhausted-like T cells | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.; Guyer, P.; Ettinger, R.A.; James, E.A. Non-Genetically Encoded Epitopes Are Relevant Targets in Autoimmune Diabetes. Biomedicines 2021, 9, 202. https://doi.org/10.3390/biomedicines9020202
Nguyen H, Guyer P, Ettinger RA, James EA. Non-Genetically Encoded Epitopes Are Relevant Targets in Autoimmune Diabetes. Biomedicines. 2021; 9(2):202. https://doi.org/10.3390/biomedicines9020202
Chicago/Turabian StyleNguyen, Hai, Perrin Guyer, Ruth A. Ettinger, and Eddie A. James. 2021. "Non-Genetically Encoded Epitopes Are Relevant Targets in Autoimmune Diabetes" Biomedicines 9, no. 2: 202. https://doi.org/10.3390/biomedicines9020202
APA StyleNguyen, H., Guyer, P., Ettinger, R. A., & James, E. A. (2021). Non-Genetically Encoded Epitopes Are Relevant Targets in Autoimmune Diabetes. Biomedicines, 9(2), 202. https://doi.org/10.3390/biomedicines9020202