Abstract
Cardiovascular disease (CVD) remains a major problem for people with type 2 diabetes mellitus (T2DM), and dyslipidemia is one of the main drivers for both metabolic diseases. In this review, the major pathophysiological and molecular mechanisms of β-cell dysfunction and recovery in T2DM are discussed in the context of abnormal hepatic lipid metabolism and cardiovascular health. (i) In normal health, continuous exposure of the pancreas to nutrient stimulus increases the demand on β-cells. In the long term, this will not only stress β-cells and decrease their insulin secretory capacity, but also will blunt the cellular response to insulin. (ii) At the pre-diabetes stage, β-cells compensate for insulin resistance through hypersecretion of insulin. This increases the metabolic burden on the stressed β-cells and changes hepatic lipoprotein metabolism and adipose tissue function. (iii) If this lipotoxic hyperinsulinemic environment is not removed, β-cells start to lose function, and CVD risk rises due to lower lipoprotein clearance. (iv) Once developed, T2DM can be reversed by weight loss, a process described recently as remission. However, the precise mechanism(s) by which calorie restriction causes normalization of lipoprotein metabolism and restores β-cell function are not fully established. Understanding the pathophysiological and molecular basis of β-cell failure and recovery during remission is critical to reduce β-cell burden and loss of function. The aim of this review is to highlight the link between lipoprotein export and lipid-driven β-cell dysfunction in T2DM and how this is related to cardiovascular health. A second aim is to understand the mechanisms of β-cell recovery after weight loss, and to explore new areas of research for developing more targeted future therapies to prevent T2DM and the associated CVD events.
1. Introduction
Type 2 diabetes mellitus (T2DM) has become a global concern. It affects 425 million of the world population, and doubling is predicted in the coming decades [1,2]. On average, 10–15% of the national health budget in western countries is directed to manage T2DM and its complications, including cardiovascular disease (CVD), the leading cause of death worldwide [3]. However, available drugs are relatively ineffective in controlling this epidemic, and there is urgent need for other means to manage T2DM and prevent development of CVD [4,5,6].
One of the major questions that remains a puzzle for people with T2DM and scientists alike is “why have I developed T2DM even though I’m not as heavy as my friends who do not have diabetes?” There are many factors that contribute to the pathophysiology of this disease, including body weight, fat mass, age, ethnicity, gender, genetics, and environmental factors [7]. However, the majority of these factors are modifiable and largely controlled by lifestyle intervention including diet and physical activities. Although it is considered a risk factor, obesity itself is not causative of T2DM [8,9]. It is known that the majority of overweight people do not develop T2DM [10]. This “metabolically healthy” phenotype is genetically determined via alleles associated with higher subcutaneous and lower ectopic fat deposition [11,12]. On the other hand, normal weight people may develop T2DM due to limited subcutaneous fat storage ability accompanied by susceptibility of insulin-producing cells within the pancreas (β-cells) to these adverse metabolic conditions, and the concept of personal fat threshold explains this phenomenon [9]. Subcutaneous adipose tissue allows metabolically well-tolerated fat storage, and this may partially explain why some people develop T2DM at normal body mass index (BMI) [9,10]. Indeed, women are generally less susceptible to T2DM and cardiovascular events, and large subcutaneous fat area in women may provide a safe zone for storage of excess triglycerides protecting the β-cells and other susceptible tissues from the harmful effects of excess lipids [7,13,14].
Genetic factors related to ability of β-cells to increase mass and function during high demand on insulin or ability of β-cells to survive the adverse metabolic conditions should be considered [15,16]. Work on animal studies support the beneficial effects of subcutaneous fat and the genetic basis of β-cell susceptibility to increased levels of glucose and fatty acids in obesity and T2DM [17,18]. In the lipodystrophy mouse model (A-ZIP/F-1), transplanting subcutaneous fat returned liver fat levels to normal and regulated blood glucose levels [19]. In addition, prefusion studies on isolated islets from the zucker diabetic fatty (ZDF) rat model have shown that simultaneous prefusion of fatty acids and glucose induced β-cell dysfunction in the homozygous rats, but not in the heterozygous littermates [18]. Genome-wide association studies (GWAS) have also highlighted the protective nature of subcutaneous fat in obesity and T2DM [12]. To date, the majority of identified polymorphisms in T2DM are related to the β-cell secretory function rather than insulin function itself [20,21]. However, it should be recognized that current studies identified less than 10% of the genetic factors that are expected to contribute to the pathophysiology of T2DM, and more genetic loci are expected to be identified in the future [15].
On the basis of recent evidence from the Diabetes Remission Clinical Trial (DiRECT), weight loss to treat T2DM is currently incorporated into the National Health System (NHS) in the United Kingdom, thereby to lower CVD risk [4,5]. However, achievement and maintenance of weight loss is difficult and requires strong motivation and long-term support for adherence to dietary conditions and prevention of weight regain following weight loss. In addition, this approach does not suit everybody, including those with normal body weight. There is a pressing need to develop more targeted strategies for remission of T2DM without the requirement for major weight loss.
“Lipotoxicity” is one of the most widely accepted hypotheses to explain the underlying mechanisms of β-cell dysfunction in T2DM [16,22,23,24]. Maintenance of normal lipid homeostasis occurs via crosstalk among many organs of the body including the liver, the pancreas, and adipose tissues. Figure 1 is a schematic hypothetical representation of the lipid-related factors that may contribute to the pancreas function in T2DM. This review outlines and discusses the major pathophysiological and molecular mechanisms of β-cell dysfunction and recovery in T2DM in the context of dyslipidemia and cardiovascular health. Additionally, it will highlight new areas for future research to develop novel therapies for T2DM. Whether β-cell uptake of these toxic fatty acids and lipid intermediates delivered by lipoproteins could be blocked at early stages to preserve β-cells and maximize their survival requires investigation. Alternatively, novel therapeutic strategies are needed that enhance diversion/depletion of excess energy intake or healthy expansion of adipose tissues without the need to upregulation of the major lipogenesis and lipoprotein export pathways.
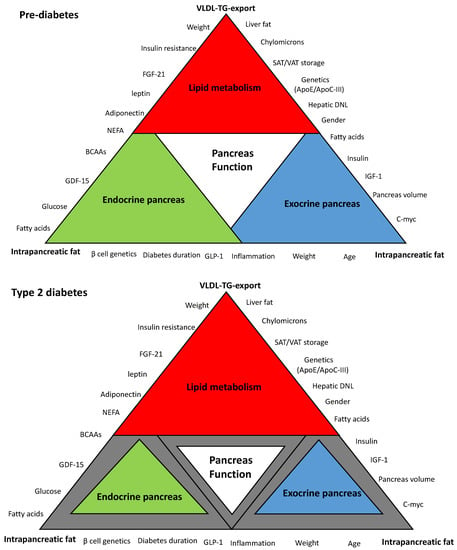
Figure 1.
Schematic hypothetical diagram of the interplay between hepatic very low density lipoprotein triglyceride (VLDL-TG) export and intrapancreatic fat in type 2 diabetes. Pancreas function is accomplished through the synergy between endocrine and exocrine compartments. Abnormalities in lipid metabolism are the drivers of metabolic events that affect overall pancreas structure and function. The extent of each colored triangle represents the degree of function in that parameter (gray color means loss of function). Lipid-driven changes that cause dysfunction of β-cells in type 2 diabetes mellitus (T2DM) and the loss of the acinar cells mass are likely to be related to hepatic VLDL-TG export and increase in intrapancreatic fat. β-cell: Beta cell; VLDL-TG: very low density lipoprotein triglyceride; FGF-21: fibroblast growth factor 21; NEFA: non-esterified fatty acids; BCAAs: branched-chain amino acids; GDF-15: growth differentiation factor-15; GLP-1: glucagon-like peptide-1; C-myc: cellular myelocytomatosis oncogene; IFG-1: insulin-like growth factor-1; DNL: de novo lipogenesis; ApoE: apolipoprotein E, ApoC-III: apolipoprotein C-III; SAT: subcutaneous adipose tissue; VAT: visceral adipose tissue.
2. Altered Lipid Metabolism and Etiology of T2DM
Excess calorie intake and ectopic fat deposition are major determining factors for the pathophysiology of T2DM [25,26,27,28]. Obesity itself, defined as a fixed cut-off of body mass index (BMI), is not causative factor [8,9], and overweight people free from diabetes may have a “metabolically healthy” phenotype with safe fat storage [12,29]. On the other hand, those who develop T2DM at lower BMI may have (i) limited storage capacity or disordered adipose tissue function, (ii) varied susceptibility to toxic lipid metabolites, and (iii) failure to increase β-cell mass appropriately during adipose tissue expansion and increased demand on insulin [15,29,30].
In T2DM, lipid metabolism is abnormal, directly related to overproduction of very low density lipoprotein (VLDL) by the liver [31,32,33]. This is accelerated through expression of transcription factors that activate lipogenesis genes including the carbohydrate response element binding protein (ChREBP) and sterol regulatory element-binding protein 1c (SREBP1c), which are activated by glucose and insulin, respectively [34,35]. Therefore, the rates of hepatic de novo lipogenesis (DNL) rise substantially in T2DM under elevated levels of glucose and insulin [34,36,37].
Liver function is central for regulation of lipid metabolism through export of fat in lipoprotein (VLDL-TG), and uptake of free fatty acids from circulation [31]. However, this mechanism is impaired in T2DM [31,32]. As the liver fails to maintain the balance between fat uptake and delivery, excess fat will be stored ectopically when subcutaneous depots are unable to accommodate more triglycerides. Thus, non-alcoholic fatty liver disease (NAFLD) is common, and directly involved in the pathogenesis of T2DM [38,39,40,41]. Excess fat will not only impair liver function in regulating blood glucose levels, but will also spillover to other ectopic sites, including the pancreas and muscles, interfering with β-cell function and cellular insulin signaling. Using magnetic resonance imaging (MRI) and intralipid infusion techniques, we have reported elevated levels of liver fat and major increase in hepatic VLDL-TG production in T2DM, and that this was normalized after remission of diabetes [26,27,42] (Figure 2). If DNL is the driver of excess liver fat accumulation and hepatic VLDL-TG production, it would be expected to decrease during remission of T2DM, and this would possibly by a target for the future prevention and remission programs.
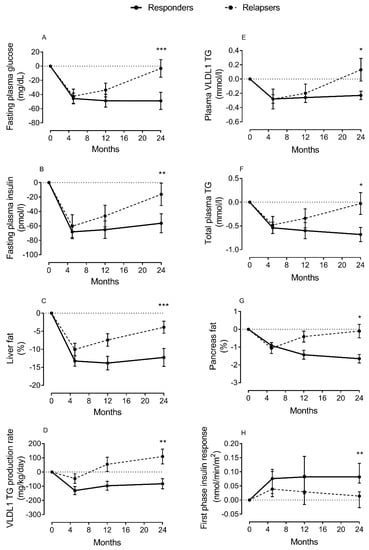
Figure 2.
Change in lipid parameters and β-cell function during remission and relapse of type 2 diabetes. Change from baseline in fasting plasma glucose (A), fasting plasma insulin (B), liver fat (C), hepatic VLDL1-TG production (D), fasting plasma VLDL1-TG (E), total plasma triglycerides (TGs) (F), intrapancreatic fat (G), and β-cell function (H) at 5 months (responders n = 38; relapsers n = 13), 12 months (n = 28/n = 13, respectively), and 24 months (n = 20/n = 13, respectively). Responders are presented as a solid black line and relapsers as a dashed line. The dotted line is the gridline at y value = 0. Paired data between baseline and each time point are presented. Data are presented as mean ± SEM except for first-phase insulin (median with IQ range) vs. 5 months in relapsers: * p < 0.05, ** p < 0.01, *** p < 0.001. Figure is presented with permission [43].
One other important function of the liver is uptake and clearance of lipoprotein remnants from the blood, which results from the catabolism of triglyceride-rich lipoproteins. This includes removal of chylomicron remnants, intermediate density lipoprotein particles (IDL), and the highly atherogenic low-density lipoprotein particles (LDL) from circulation. The process is mediated by certain receptors on the hepatocytes and controlled by the function of several apolipoproteins including apoB, apoE, and ApoC-III. Therefore, liver function is a major determining factor for CVD. The underlying mechanisms of how weight loss reverses fatty liver and achieves remission of diabetes remain largely unclear [10,27,43]. Recent data from rodent studies highlighted the role of diacylglycerol (DAG) in activation of the ε isoform of hepatic protein kinase C (PKCε), which impairs insulin function, and weight loss was reported to reverse this process [44,45,46]. In addition, long-chain saturated fatty acids have also been reported to activate the toll-like receptor 4 (TLR-4) and generate toxic ceramides that inhibit insulin signaling [46,47,48].
Insulin is the master regulator of lipid metabolism; it is known to inhibit lipolysis to maintain non-esterified fatty acid (NEFA) levels by suppressing the function of hormone-sensitive lipase (HSL) [49]. Insulin is a regulator of hepatic VLDL production indirectly via downregulating transcription of the ApoC-III and the microsomal triglyceride transfer protein (MTP) [50]. It is also a regulator of the transcription factor forkhead box protein (FoxO1), which in addition to regulation of gluconeogenesis upregulates the expression of ApoC-III and MTP and thereby enhances lipidation and secretion of VLDL by the liver [51,52]. Expression of ApoC-III in the β-cell of mice impaired insulin secretion [53], whereas FoxO1 expression protected β-cell from adverse metabolic conditions [54]. Furthermore, proprotein convertase subtilisin kexin type 9 (PCSK9) may have effects in lipid metabolism and β-cell function in T2DM, and use of anti-PCSK9 monoclonal antibodies has a potential to prevent new-onset diabetes in the future, although the evidence is currently lacking [55].
Whether β-cell dysfunction in T2DM is a result of toxic lipids or related to other effector proteins produced by the β-cell itself or other organs remains an open question, and this is currently an active area of research. Hypoxia and oxidative stress are known in obesity and T2DM [56]. This new redox environment could be the initiation factor in altering lipoprotein metabolism and generation of reactive lipid species, which in turn alter adipose tissue function and the biology of β-cell to survive these lipotoxic conditions. Investigation of the biochemical changes of lipoproteins and related lipid products during development and remission of diabetes may therefore lead the way to new therapeutic targets. In this regard, proteomics and metabolomics studies using advanced mass spectrometry techniques will reveal the underlying changes in the biochemistry and function of lipoproteins and lipid-related molecules.
3. Lipotoxicity and β-Cell Dysfunction
Loss of β-cell function is critical factor to the pathogenesis of T2DM. Despite debate about the terminology, “lipotoxicity” remains the most widely accepted hypothesis to explain β-cell dysfunction in T2DM [16,23,24,57]. This review is directed towards giving an overview of the evidence in favor of the adverse effect of lipids on the pancreas and β-cell function, and does not cover “lipotoxicity” or “glucotoxicity” in detail. For more detailed information, other recent reviews cover the topic at cellular and molecular levels [16,23,24,58,59,60].
At normal physiological conditions, fatty acids are known to stimulate insulin secretion from the pancreatic β-cells [61,62]. Therefore, saturated fatty acids that are a major component of the modernized diet are potentially the triggers for excessive basal insulin. This in turn is permissive for a cluster of major metabolic abnormalities including insulin resistance, NAFLD, and dyslipidemia [59]. Various hypotheses to explain the glyco-lipotoxic effects on β-cell function in T2DM have been postulated, including apoptosis, endoplasmic reticulum (ER) stress, oxidative stress, inflammation, mitochondrial dysfunction, autophagy, and de-differentiation [16,22,23,63,64]. However, the precise mechanisms of how toxic lipids can induce stress and eventually dysfunction of the β-cell remain critically important to be established [7,22].
Most of the available data on “lipotoxicity” are based on in vitro studies of β-cell lines or isolated islets, and most confirmed the deleterious effects of palmitate in causing ER stress or apoptosis when incubated with β-cells at high concentration [65,66]. Incubation of β-cells with palmitic acid (C16:0) combined with either oleic acid (C18:1) or arachidonic acid (C20:4) prevented the cellular damage that was induced by palmitic acid alone [65,67,68]. On the other hand, incubating β-cells with arachidonic acid enhanced β-cell proliferation and increased insulin secretion in cultured cell lines and β-cells [67,68]. It is important to consider that all these in vitro studies used a high concentration of fatty acids that are not encountered under physiological conditions, and hence there is no concrete in vivo evidence yet of the proposed lipotoxic effect of fatty acids in humans [57]. Under physiological conditions, β-cells are exposed to a mixture of nutrients including glucose, fatty acids, and amino acids, and thus the term “nutri-stress” that was proposed recently by Prentki et al. is more appropriate instead of “lipotoxicity” or “glucolipotoxicity” [23].
Fatty acids have been reported to contribute to the β-cell dysfunction on the basis of the number of carbons in the fatty acyl chain and the degree of saturation. Saturated fatty acids with long chain (i.e., C16:0 palmitic acid) have been reported to induce cell death or apoptosis whereas unsaturated fatty acids (i.e., C18:1 oleic acid) had opposing effects [59,60,68,69]. In the support of these cellular findings, it has been demonstrated that palmitate-induced ER stress was modulated by the expression of stearoyl-CoA desaturase and Elovl6 in rodents [70,71].
In vivo work on β-cell lipotoxicity was pioneered over 30 years ago by Roger H. Unger in rodents [18,72]. In the ZDF rat, triglyceride content of the islets increased 10 times during development of T2DM, a few weeks before development of hyperglycemia, and this correlated strongly with circulating fatty acids. In addition, this rise in islet fat associated with absence of glucose-stimulated insulin secretion and low expression of GLUT-2 of the β-cells [18]. Using the same model, it was found that infusion of intralipid and glucose decreased β-cell function in older but not in younger animals [73]. However, it is important to consider that intralipid mainly contains unsaturated fatty acids that were reported not to have toxic effects on β-cells.
The evidence of lipotoxic effect of fatty acids is less significant in humans, although there is a general agreement about the synergic effects of glucose and fatty acids on β-cell dysfunction in T2DM [23,57,74]. “Metabolic inflexibility” of the cell to switch between glucose and fatty acid metabolism is well known [75], and this is supported by our recent indirect calorimetry data showing a decrease in lipid oxidation accompanied by an increase in glucose oxidation after remission of T2DM [27]. It was reported recently that palmitic acid is not effective fuel for β-cell in rodents [76,77], and this may partially explain β-cell dysfunction in T2DM. Some observational and lipid infusion studies have reported association between NEFA and β-cell function in humans, whereas others found no such evidence [23,57,78,79,80]. Interestingly, using positron emission tomography (PET), researchers found that fatty acid uptake by the pancreas was higher in obese individuals compared with normal weight controls, and this was associated with lowered glucose uptake and blood flow, which was associated negatively with markers of β-cell function [81].
It is important to recognize that the regional level of NEFA is under tight control of insulin, and this is normally regulated by cross talk between the liver, adipose tissues, and pancreas [49,82]. In addition, circulating NEFA makes only one portion of the fatty acids encountered by the pancreas, and the concentration of NEFA in blood is low [83]. There are several other sources for β-cell uptake of fatty acids. (I) Lipoprotein lipase (LPL) and hence uptake from circulating triglyceride can regulate β-cell function [84]. (II) Adipocyte infiltration in close proximity to the pancreatic islets in T2DM and hydrolysis of triglyceride content are other sources of fatty acids for the β-cells [65,85]. (III) Formation of lipid droplet within the β-cell was found in T2DM [86], and high expression of HSL in β-cell has decreased insulin secretion after feeding mice on a high fat diet, and this was associated with lower triglyceride accumulation within the islets of the transgenic mice compared with the wild-type mice [87]. Furthermore, toxic lipid intermediates derived from fatty acid metabolism including diacylglycerides and ceramides were confirmed to cause damage to insulin signaling within the hepatocytes and myocytes [88,89], but less is known about how such toxic metabolites can affect β-cells [7,57]. Thus, the lack of correlation between plasma NEFA and β-cell dysfunction does not provide evidence against the lipotoxic effect of lipids on β-cell function.
Earlier studies claimed that apoptosis could explain loss in β-cell mass and function in T2DM [64,90,91,92]. However, there is no strong evidence to support β-cell death from either animal or human studies [23,30,93]. On the other hand, loss of specialized β-cell phenotype (de-differentiation) could be explained by glucolipotoxicity [63,94,95,96,97], and this is the most likely mechanism to explain return of β-cell function after remission of T2DM [10,98]. Under metabolic conditions of excess fat and eventually glucose, some β-cells lose their identity to become glucagon-producing α-cells [96,99]. Conclusive data confirming β-cell de-differentiation are limited, especially in humans [63,96]. More work is needed to confirm whether de-differentiation/re-differentiation are the main underlying molecular mechanisms of β-cell dysfunction and recovery in T2DM.
4. The Link between Lipoprotein Export and β-Cell Dysfunction
Is there clear evidence of the link between lipoprotein export and “lipotoxicity” or dysfunction of the β-cell? The “Twin Cycle” hypothesis postulates that hepatic lipoprotein export is the upstream pathway that delivers excess fat to the pancreas and leads eventually to β-cell dysfunction [28]. However, until now, there has been no direct evidence to support the idea that VLDL-TG export is the source of fat buildup within the pancreas, and this remains a hypothesis (Figure 1). Indirectly, we have shown that remission of diabetes is associated with fall in hepatic VLDL-TG export, and return to diabetes status has been associated with increase in plasma VLDL-TG levels [43]. Direct uptake of VLDL particles by the β-cells was reported in humans, and expression of LPL within the β-cell was found in mice and affected insulin secretion [84,100]. Isolated islets from mice lacking the LDL receptor showed lower LDL uptake and more survival rate than mice that expressed the LDL receptor [101]. In addition, it was reported that LPL is expressed in the capillaries of pancreatic islets, and this ensures delivery of postprandial fatty acids from chylomicrons to the β-cells [83]. Local expression of ApoC-III within the pancreatic islets has led to β-cell failure in mice [53]. In contrast, it was found that high-density lipoprotein (HDL) expression in β-cell had a protective role against ER stress [102].
High intrapancreatic fat content and infiltration of adipocytes in close proximity to pancreatic islets are known in T2DM [65,103]. Recently, we have demonstrated that normalization in liver fat content and hepatic VLD-TG export is related to the fall in the palmitic acid component of VLDL-TG, the pathway that delivers fat to the peripheral tissues including the pancreas [43]. This fatty acid is the obligatory product of DNL and the most toxic fatty acid to the β-cell at high concentration and under prolonged exposure [69]. Interestingly, loss of β-cell function during relapse into diabetes was found to be associated with a rise in plasma levels of VLDL-TG, highly enriched with palmitic acid, and an increase in intrapancreatic fat levels (Figure 2).
Despite evidence about the role of toxic lipids on the pathogenesis of NAFLD, there are limited data on such effects on the β-cells in humans [57]. This is mainly due to the limited access to human pancreatic tissues. Unlike the liver, pancreas biopsy is a very invasive process and is not feasible in clinical practice. Available data in humans are derived from pancreas samples collected from post-mortem donated organs and excised tissues during surgery, which are not ideal conditions and could partially explain the conflicting reports [104]. There is urgency to develop non-invasive imaging techniques to study β-cells in vivo because of these obstacles, and novel PET-MRI methods using safe tracers are currently under development [105].
It has been known for a long time that the biology and structure of pancreatic islets are different in human and rodents, and this is reflected in the β-cell’s ability for adaptation to adverse metabolic conditions [106,107,108]. Therefore, recent work should focus on human studies considering these major differences between species. Recent RNA-sequencing data from human islets showed that saturated fatty acids induced β-cell stress independent of the major inflammatory pathways [109]. A recent study reported that higher expression of the cluster differentiation 36 (CD36) receptor is associated with defective insulin secretion in β-cells from obese donors with T2DM [110]. CD36 is a transporter protein that determines fatty acid uptake by the β-cells and could be a potential target to block β-cell uptake of these toxic lipid products. In addition, this CD36 receptor was found to be localized on the insulin granules in human β-cells and found to control fatty acid stimulation of insulin secretion [111]. Further work is required to identify potential toxic lipid species and the underlying mechanisms of their toxic effects on β-cell function to inform alternative treatments for T2DM.
5. Adipokines and Lipid Related Markers
Lipid homeostasis in humans is maintained by high coordination between the liver, the pancreas, and the adipose tissues. Although it has been considered for long time as a storage depot, the metabolic function of adipose tissues has gained more interest in recent years after the discovery of leptin and adiponectin [112]. Adipose tissue function should therefore be taken into consideration when discussing β-cell dysfunction, T2DM, and CVD.
Leptin is predominantly produced by adipocytes, and plasma levels reflect total body fat mass; thus, leptin level is always higher in women than men at any given BMI [113,114]. Leptin regulates fat storage as well as appetite and has been associated with both glucose- and lipid-lowering effects [18,19,20]. Adiponectin, another adipocyte-derived hormone, has an antidiabetic effect [21,22]. In contrast to leptin, adiponectin level is negatively correlated with adiposity [23]. The plasma leptin to adiponectin ratio is considered a marker of atherogenicity in T2DM [115,116]. It is known that leptin level is elevated in obesity and T2DM, and resistance to leptin action is widely accepted [117,118]. Dietary weight loss has been reported to cause decrease in leptin and increase in adiponectin levels [119,120,121,122].
Inflammation in adipose tissue is likely a consequence of excess fat accumulation. However, the role of inflammatory cytokines on adipose tissue biology is uncertain. Whether or not inflammatory cytokines contribute to β-cell dysfunction and CVD development requires further investigation, and this is beyond the scope of this review [123,124]. Low-grade inflammation was found to be associated with C16:0 ceramide, and lowering plasma levels of this lipid species was reported as a potential therapy for T2DM [125,126]. However, saturated fatty acids were found to induce ER stress in β-cells independent of the major inflammatory pathways [30,31].
It has been suggested that lipids and branched-chain amino acids (BCAAs) could have a synergic effect in causing insulin resistance in T2DM, but little is known about their effect on β-cell function [127]. Several studies have reported elevated plasma BCAAs in obesity and T2DM [128,129]. In rodents, BCAAs were reported to stimulate insulin secretion and activate the mTORC1, which is related to β-cell mass and function [130]. mTORC1 is a negative regulator of autophagy [131], and calorie restriction modulates autophagy, a process that known to regulate lipid metabolism [132,133,134,135]. There are conflicting reports as to whether the change in BCAA level after weight loss in human is related to weight loss per se or to diabetes status [134,135].
The growth and differentiation factor 15 (GDF-15) is related to nutritional stress, and it is elevated in T2DM and CVD [136,137]. In addition, the fibroblast growth factor 21 (FGF-21) is secreted by the liver and is highly expressed in the pancreas. FGF-21 is strongly associated with obesity and T2DM as a regulator of lipid and glucose metabolism [138,139,140]. It selectively promotes glucose uptake in adipocytes, and thereby complements the function of leptin and adiponectin in promoting TG storage in adipocyte and fatty acid oxidation in other tissues [140,141,142]. FGF-21 levels are elevated in patients with T2DM, possibly suggesting resistance to its function [117,138,143]. In rodents, FGF-21 stimulates adiponectin, which exhibits ceramidase activity, degrading toxic ceramides and enhancing insulin sensitivity [144,145,146]. In addition, hepatic expression of FGF-21 was reported to enhance the cellular process that leads to browning of adipose tissues [147,148]. Notably, brown adipose tissues, which contribute to regulation of body metabolism and energy expenditure via oxidation of glucose and lipids, are more abundant in women [149]. Furthermore, it has been shown that FGF-21 shifts VLDL-TG uptake from white to brown adipose tissues in obesity [150]. Interestingly, FGF-21 was found to regulate hepatic VLDL-TG secretion in mice via accelerating adipose tissues uptake mediated by CD36 and LPL functions [150].
Identification of generic biomarkers for cell stress and differentiation would be useful to understand the underlying mechanism of β-cell failure and recovery in T2DM in light of the lack of β-cell-specific dysfunction markers [136,138,139]. Further work is needed to understand the exact functions of GDF-15, FGF-21, and BCAAs in relation to lipid metabolism and β-cell function in humans.
6. Intrapancretic Fat and Pancreas Morphology
Although the pancreas is the most important organ for diabetes, studies on it are limited. Clinical and observational studies of the pancreas have naturally focused on islet function itself, but the relevance of acinar cells to endocrine function has rarely been considered [151,152,153,154]. The pancreas volume is small in T2DM, with marked irregularity in pancreas borders [155,156]. Acinar cell mass reflects total pancreas volume with islet and ductal systems contributing ≈5%. In addition, fat content of the pancreas is moderately elevated in T2DM [25,27,157], correlating negatively with pancreas volume [155]. Recently, we have confirmed that normalization of intrapancreatic fat content returned β-cell function with normalization of maximal β-cell capacity [158].
Studies on post-mortem pancreas of people with T2DM have shown fibrosis in exocrine tissues associated with decline in β-cell and increase in α-cell mass [159]. In ZDF rat, fat replacement of the acinar cells developed into fibrosis [160], possibly leading to destruction of the islets and β-cell dysfunction [153,160,161,162]. Furthermore, trans-differentiation of the acinar cells into adipocytes occurs in mice during ageing, regulated through expression of c-Myc transcription factor [163]. Whether these reported changes in acinar cells are related or secondary to loss of β-cell function is an important question. Inadequate insulin secretion during development of T2DM could possibly explain the observed atrophy of pancreas volume due to lack of trophic effect of insulin on pancreatic tissue [164]. Notably, pancreas volume is small in people with type 1 diabetes (T1DM) when insulin secretion is absent [165,166]. The trophic effects of insulin at high concentrations such as experienced by pancreatic tissues following a meal could be considerable [164,167]. Insulin-like growth factor 1 (IGF-1) level is low in T1DM, ageing, and T2DM [168,169,170,171]. IGF-1 receptor shares high homology with insulin receptor, and cross-reactivity of insulin with IGF-1 receptor bring about tissue growth effects at a high concentration of insulin. It is possible that the lack of both IGF-1 and insulin are involved in the loss of acinar cell mass in T2DM. Further work on the underlying molecular mechanisms, particularly the role of IGF-1 and c-Myc, is required to understand the relevance of pancreas morphology to the pathogenesis of T2DM.
It has been reported that lipid droplets are more common within acinar cells of human donors with T2DM [86], and higher expression of the CD36 fatty acid transporter protein was associated with defective insulin secretion [39]. Recently, we have demonstrated that remission of T2DM and recovery of β-cell function were associated with gradual normalization of the pancreas morphology (Figure 3) [172]. The increase in pancreas volume after removal of excess fat suggests that this could be mediated by high post-meal insulin surges and other factors. Regeneration of pancreas tissues is known, and this may explain the observed increase in pancreas volume during remission of diabetes [173]. Whether the underlying mechanisms of toxic effect of fatty acids are similar between β-cells and acinar cells remains to be elucidated in human. Ultimately, this could lead the way to novel strategies to maintain β-cell function and preserve normal pancreas health.
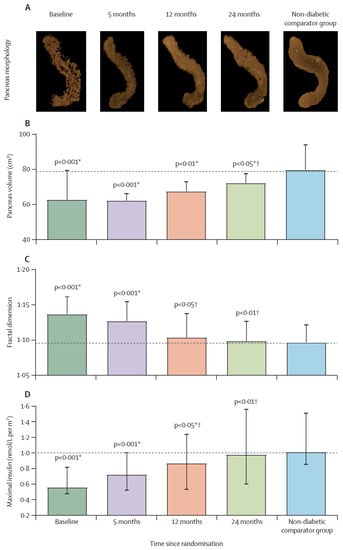
Figure 3.
Restoration of pancreas morphology and β-cell functional capacity after 2 years of remission of type 2 diabetes. Surface-rendered image of pancreas morphology in a representative responder (A), and pancreas volume (B), fractal dimension (C), and maximal insulin secretion (D) in responders compared with non-diabetic controls at baseline, 5 months, 12 months, and 24 months. Horizontal dashed lines indicate the level for non-diabetic controls. Data paired with baseline at each timepoint are presented as mean (SD) for pancreas volume and fractal dimension and median (IQR) for insulin secretion. * Responders versus non-diabetic comparator group. † Responders versus baseline. Figure is presented with permission from The Lancet Diabetes and Endocrinology [172].
7. Lipoprotein Metabolism and CVD Risk in T2DM
People with T2DM face a considerably increased risk of CVD due to disordered lipoprotein metabolism [32,174,175]. The risk of atherosclerosis has historically been attributed to the rise in hepatic secretion of triglyceride-rich VLDL, decrease in HDL cholesterol, and the rise in the small, dense LDL particles [175]. However, ApoB has been considered a more useful factor in determining CVD risk than the traditionally used LDL cholesterol levels [176]. The risk of incident T2DM has been reported to be determined by the size of the lipoprotein particles rather than lipid content and hence by the dynamics of lipoprotein secretion and clearance by the liver [177]. There is currently increased interest in the effect of lipoprotein (a), which is found to be a risk factor for CVD [178,179].
Apolipoproteins (ApoB/ApoE) are important for the assembly and clearance of lipoproteins [50,180,181]. Lack of clearance of lipoproteins remnants was reported to be related to interference with the binding of ApoB/ApoE receptors via high hepatic expression of ApoC-III [182,183]. However, it was reported recently that ApoC-III promotes LPL activity independent of ApoE-mediated clearance of lipoproteins [184]. There are conflicting reports as to whether plasma level of ApoE can predict CVD [185,186]. However, it should be recognized that not all the LDL particles in circulation carry ApoE, and thus ApoE level does not necessarily reflects the atherogenic nature of the whole lipoprotein population. Polymorphisms of ApoE gene and ApoC-III were reported to increase the risk of developing T2DM and CVD, which requires further investigation [181,187]. In addition, high expression of ApoC-III within the pancreatic islets caused β-cell failure in mice [53]. Downregulation of ApoC-III expression could therefore be used as a therapeutic target to enhance lipolysis, decrease β-cell damage, and prevent CVD risk.
Women have a lower risk of T2DM and CVD than men [13,14]. However, once T2DM develops, the risk rises to a very high level in men [188,189]. Plasma level of VLDL-TG is higher in men than in women [190], pointing to the difference in hepatic lipoprotein export and clearance. It is likely that adequate subcutaneous fat storage capacity may permit safe clearance of liver exported fat in women. However, once these stores become inflamed or reach maximal capacity to safely store excess fat, women are especially prone to excess lipid damage. Higher levels of leptin and adiponectin in women are consistent with the protective nature of these markers in developing T2DM and CVD [17,113,149].
The recent guidelines of the European Association for the Study of Diabetes (EASD) and the European Society of Cardiology (ESC) recommend statins for lowering lipid profile and prevent CVD mortality in patients with T2DM in both sexes, although the potential differences between women and men were highlighted [191].
8. Mechanisms of Remission of T2DM after Weight Loss
Weight loss studies have highlighted the likely deleterious effect of excess fat on β-cell function [25,26,27]. In T2DM, hepatic lipoprotein export is associated with high intrapancreatic fat accumulation and loss of β-cell function [26,27,43]. Decrease in both hepatic and intrapancreatic fat is a prerequisite for diabetes remission with ultimate dependence on β-cell ability to recover function after removal of this metabolic stress [27,43]. Furthermore, weight regain and loss of remission have been found to be associated with a major rise in hepatic VLDL-TG production and plasma VLDL-TG concentration (Figure 2). These data are consistent with the “Twin Cycle” hypothesis that was proposed to explain the etiology and reversibility of T2DM [28,192]. This specific enrichment of palmitic acid, known to be toxic to the β-cells, within the lipoprotein exported from the liver is consistent with a causative effect on β-cell dysfunction. The degree of weight loss, lipoprotein export, and weight stability were the major predictors for remission [43,193]. Other studies that investigate the molecular mechanisms of long-term body weight maintenance after weight loss are required. RNA sequencing of adipose and skeletal muscle biopsies showed that acylcarnitine species can determine weight regain after weight loss [194]. This result is consistent with our recent finding of the rise of palmitic acids within the VLDL-TG during weight regain and loss of remission [43], and also in agreement with the regulatory function of CerS6-dependent C16:0 ceramide on weight gain in mice [47]. Collectively, these data highlight novel approaches for the treatment of obesity and T2DM in the future.
Bariatric surgery has long been known to be effective in normalizing blood glucose and achieving remission of T2DM [195]. We have shown that the underlying physiology of diabetes reversal after surgery is the same as dietary weight loss and related to removal of ectopic fat from the liver and the pancreas [157]. The general similarity of mechanisms has also been reported by others [196,197]. Although minor improvement in β-cell function happen within a week after surgery, long-term normalization requires a longer time to remove excess fat from the pancreas [157,198]. The increase in post-meal levels of glucagon-like peptide-1 (GLP-1) immediately after surgery is unlikely to contribute to the observed rapid normalization of fasting plasma glucose. It is likely to be related to surgery procedures themselves, given that GLP-1 levels do not change after dietary weight loss despite similar glycemic improvement [119,199]. In contrast, the level of ghrelin increases following dietary weight loss but not after bariatric surgery [119,193,195]. Taken together, neither GLP-1 nor ghrelin causes remission of diabetes following weight loss, and the rise in the levels of these hormones reflects counter-regulatory mechanisms to the effects of surgery and hunger, respectively.
Long-term nutrient overload leads to loss of β-cell specialized function in secretion insulin, and removal of the lipotoxic environment will decrease uptake of the toxic lipids and restore β-cell function [22,28]. The demonstrated return of β-cell function after removing the metabolic stress following weight loss is not compatible with β-cell death or “apoptosis” [10,24,98]. Regeneration of β-cell is another process that may account for recovery β-cell mass after remission, and some drugs have been reported to have such proliferative effects. However, evidence of β-cell replication or proliferation is lacking in adult humans [108,200,201].
An appropriate diet is critical for DNL regulation and metabolic health. A diet that contains a high level of carbohydrates is known to increase DNL rate, whereas a ketogenic diet has opposing effects [202,203]. A low-calorie diet that restricts methionine was shown to delay diabetes development in mice [204]. In addition, BCAA restriction improves metabolic health in both mice and humans [205]. Given the insulin secretion stimulatory effect of BCAAs on β-cells [130], the high levels of BCAAs observed in obesity and T2DM may suggest a deleterious long-term effect or resistance to their function similar to what happens to FGF-21 in obesity [128,129,138]. Restriction of BCAAs was reported recently to worsen ketogenesis and cause oxidative dysfunction of the mitochondria in mice [206]. A ketogenic diet could be a useful approach to burn excess calories and harness less energy by the cell theoretically, although randomized controlled trials have shown only a minor effect [207]. However, more studies should be carried out to assess the adverse metabolic effects of this extreme diet including high-circulating NEFA and their potential lipotoxic effects [203].
The β-cell workload hypothesis has also been proposed to explain dysfunction and decline in β-cell mass in T2DM [64,208]. This is essentially based on the high demand on β-cells to increase insulin secretion in obesity, leading eventually to β-cell death or dysfunction. The hypothesis also explains the ethnicity difference between Asians and Caucasians in terms of the discrepancy in β-cell capacity to increase mass during obesity and T2DM. Recent reports on remission of diabetes and return of β-cell function following weight loss fits well with the workload hypothesis if discussed in the context of “lipotoxicity” and dysfunction of β-cell rather than death [10,158]. Calorie restriction not only lifts the chronic nutrient burden encountered by the β-cells, but also will remove the stimulus to produce excess circulating insulin. This is critical in triggering the cellular processes that favor use of energy rather than storage via downregulation of lipogenesis genes and normalization of hepatic lipid metabolism. Further studies to understand the exact mechanisms of β-cell recovery after weight loss are critically important for sustained remission of T2DM.
9. Novel Directions to Study Pancreas and Lipid-Mediated β-Cell Dysfunction
At normal physiological conditions, palmitic acid stimulates β-cells in secreting insulin [61,62,209]. However, prolonged exposure of β-cells to this stimulatory effect will cause stress, which is the trigger of lipotoxicity or β-cell dysfunction [23,59]. It is possible that chronic stimulation of β-cells indirectly leads to β-cell dysfunction and development of CVD by altering hepatic lipoprotein metabolism and the biology of adipose tissue. Therefore, healthy β-cell function is central for prevention of T2DM and maintaining normal cardiovascular health [7]. Current pharmacological therapies such as sulfonylureas that increase insulin secretion are potentially harmful to β-cells in the long-term. Future therapies should be targeted to remove the lipotoxic environment and hence to support β-cells to return to normal function.
At diagnosis of T2DM, the majority of β-cells (60–70%) have already lost function. Although the process takes almost a decade, there is currently no method to non-invasively assess the progressive deterioration of the pancreas during development of T2DM prior to rise of blood glucose levels [105,210]. Our work has shown that dysfunction of β-cell and decline of pancreas volume are associated with high levels of intrapancreatic fat and increase in diabetes duration [26,155]. It would be reasonable to postulate that the abnormal pancreas morphology in T2DM is driven by the same mechanism that causes β-cell dysfunction (“lipotoxicity”), and hepatic DNL is a likely initiating factor [43]. The DNL pathway could be modulated at early stage during progression of T2DM.
Although it is widely accepted, the “lipotoxicity” hypothesis to explain β-cell dysfunction is not yet proven, especially in human studies [57]. Further research is required to identify the potential toxic lipid species or combinations and to determine how exactly they induce cellular dysfunction. Access to human pancreatic tissues is critical, especially when advanced imaging techniques are utilized [211]. Advanced imaging mass spectrometry (IMS) techniques on precision cut pancreatic slices would be very useful for in situ localization of lipid species within the pancreatic tissues in close proximity to the islets. In this regard, the Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) system could be useful in getting a full profile of lipid species at subcellular resolution [212,213]. It could be used to determine the chemical structure of lipids within the lipid droplets inside the adipocytes, and within individual β-cells or acinar cells. MALDI (TOF/TOF) imaging could also be used for co-localization of lipid binding protein receptors (CD36) [214]. IMS data could be confirmed by high-resolution electron and confocal microscopy imaging. Flow cytometry imaging, metabolimetry, and live cell imaging are other complementary techniques that could also be useful [215,216,217]. In addition, lineage tracing studies could be illuminating for identification the origin of β-cells, α-cells, and adipocytes within the pancreatic tissues [218].
Excessive expansion of adipose tissues changes the biology of adipocytes, decreasing their secretory ability of adipokines. This unhealthy situation will activate pro-inflammatory macrophages that produce inflammatory cytokines including tumor necrosis factor alpha (TNFα) and interleukin 6 (IL-6) [219]. Non-invasive imaging techniques that can detect early signs of adipose tissue inflammation could be useful in assessing the capacity of fat storage as a risk factor for developing T2DM and CVD. In addition, understanding the molecular basis of adipose tissue expansion is critical for modeling their function. The peroxisome proliferator-activated receptor-γ (PPARγ) is a transcriptional regulator of adipocyte maturation and an essential factor mediating the function of these cytokines and regulation of insulin secretion [220,221]. Perilipin 5 (PLIN5), which is another transcriptional regulator of adipocyte, was found to regulate insulin secretion in the islets of humans and mice in a cAMP-dependent manner, and to prevent the lipotoxic damage to β-cells [222,223]. It is possible that excessive insulin secretion leads indirectly to β-cell dysfunction and eventfully development of CVD by altering hepatic lipoprotein metabolism and promoting unhealthy expansion of adipose tissues.
A combination of human and animal studies is required to understand the toxicity of lipid species on β-cell function and the role of the above transcription factors on lipid metabolism and β-cell survival. Ultimately, this could lead the way to novel strategies (Figure 4) for prevention and remission of T2DM other than the current blunt weight loss approach. Early actions are required to monitor pancreas health through the development of novel in vivo imaging techniques. In addition, localization of toxic lipid species is essential to understand the underlying mechanisms via developing advanced ex vivo imaging methods at cellular and subcellular levels. Furthermore, identification of the potential oxidative post-translational modifications of lipoprotein and lipid-related moieties during development of T2DM will open new windows for advanced targeted therapies in the future.
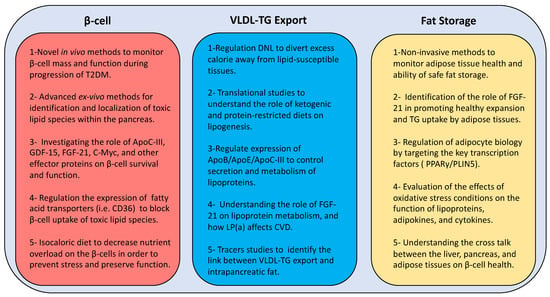
Figure 4.
New directions of research for developing novel therapies to manage type 2 diabetes. T2DM: type 2 diabetes mellitus; GDF-15: growth differentiation factor 15; FGF-21: fibroblast growth factor 21; C-myc: cellular myelocytomatosis oncogene; β-cell: beta cell; CD36: cluster differentiation 36; DNL: de novo lipogenesis; ApoB: apolipoprotein B; ApoE: apolipoprotein E; ApoC-III: apolipoprotein C-III; LP(a): lipoprotein (a); VLDL-TG: very low density lipoprotein triglyceride; PPARγ: peroxisome proliferator-activated receptor-γ; PLIN5: perilipin 5.
10. Conclusions
The pancreas is a key regulator of hepatic lipids, and healthy pancreas function is central for regulation of whole-body metabolism. The decline in acinar cell mass and loss of β-cell function are two major pathological process associated with excess intrapancreatic fat in T2DM. Therefore, early non-invasive diagnosis methods are useful for maintaining pancreas health and prevention of T2DM and related cardiovascular complications.
The classical definition of “lipotoxicity” should not be restricted to the deleterious effects of fatty acids. It should be extended to include the role of other lipid species including lipid-related markers and signaling molecules. Despite indirect evidence of the lipotoxic effects on β-cell function in T2DM, more work is required to identify the nature and function of the potential toxic lipid species. Advanced in situ imaging will be invaluable to identify and colocalize lipid species at subcellular resolution. This will inform developing novel therapeutic strategies via use of neutralizing antibodies or ligands that can decrease or regulate cellular uptake of these toxic products. For example, regulation of the expression of the CD36 transporter would be a promising approach in the future to block β-cell uptake of palmitic acid and related toxic intermediates.
Novel therapeutic strategies for remission of T2DM should focus on the key pathophysiological pathways that are regulated after weight loss. Effective strategies should initially decrease excess insulin and alter the lipotoxic environment. Use of insulin secretion enhancing drugs increases the metabolic burden on remaining β-cells, possibly leading to non-reversible damage. Notably, hepatic lipoprotein export pathway should be targeted to allow safe storage of excess calories. This could be achieved by dietary or pharmaceutical means via targeting the key regulatory elements (i.e., DNL, ApoC-III, LPL). Genetic, cellular, and translational studies accompanied by advanced in vivo imaging and mass spectrometry techniques will increase our knowledge in the future about the mechanisms behind unhealthy expansion of adipose tissues and β-cell dysfunction. This should be exploited to design novel strategies to promote storage of fat away from ectopic sites and decrease cellular exposure to toxic lipids. In this regard, FGF-21 has great effects in regulation the uptake of excess triglycerides and selective expansion of adipose tissues, and could potentially lead to attractive therapeutic options.
Funding
The author reports no funding associated with preparation of this review.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to thank Roy Taylor and the wider research team I worked with over the past 7 years. I am also grateful to our participants and to Diabetes UK for funding our research.
Conflicts of Interest
The author reports no conflict of interest.
References
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine 2019, 47, 22–27. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lean, M.E.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): An open-label, cluster-randomised trial. Lancet 2018, 391, 541–551. [Google Scholar] [CrossRef]
- Lean, M.E.J.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 344–355. [Google Scholar] [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A. Pathogenesis and remission of type 2 diabetes: What has the twin cycle hypothesis taught us? Cardiovasc. Endocrinol. Metab. 2020, 9, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R. Obesity and Type 2 Diabetes: What Can Be Unified and What Needs to Be Individualized? Diabetes Care 2011, 34, 1424–1430. [Google Scholar] [CrossRef]
- Taylor, R.; Holman, R.R. Normal weight individuals who develop Type 2 diabetes: The personal fat threshold. Clin. Sci. 2014, 128, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Al-Mrabeh, A.; Sattar, N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 726–736. [Google Scholar] [CrossRef]
- Ji, Y.; Yiorkas, A.M.; Frau, F.; Mook-Kanamori, D.; Staiger, H.; Thomas, E.L.; Atabaki-Pasdar, N.; Campbell, A.; Tyrrell, J.; Jones, S.E.; et al. Genome-Wide and Abdominal MRI Data Provide Evidence That a Genetically Determined Favorable Adiposity Phenotype Is Characterized by Lower Ectopic Liver Fat and Lower Risk of Type 2 Diabetes, Heart Disease, and Hypertension. Diabetes 2018, 68, 207–219. [Google Scholar] [CrossRef]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J.; et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 2017, 49, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.; Walker, J.J.; Colhoun, H.M.; Leese, G.P.; Lindsay, R.S.; McKnight, J.A.; Morris, A.D.; Pearson, D.W.; Petrie, J.R.; Philip, S.; et al. Do men develop type 2 diabetes at lower body mass indices than women? Diabetologia 2011, 54, 3003–3006. [Google Scholar] [CrossRef] [PubMed]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Méneur, C.; Bernal-Mizrachi, E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Asp. Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Onodera, T.; Scherer, P.E. Lipotoxicity and β Cell Maintenance in Obesity and Type 2 Diabetes. J. Endocr. Soc. 2019, 3, 617–631. [Google Scholar] [CrossRef]
- Tran, T.T.; Yamamoto, Y.; Gesta, S.; Kahn, C.R. Beneficial Effects of Subcutaneous Fat Transplantation on Metabolism. Cell Metab. 2008, 7, 410–420. [Google Scholar] [CrossRef]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-Cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-Beta-Cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef]
- Kim, J.K.; Gavrilova, O.; Chen, Y.; Reitman, M.L.; Shulman, G.I. Mechanism of Insulin Resistance in A-ZIP/F-1 Fatless Mice. J. Biol. Chem. 2000, 275, 8456–8460. [Google Scholar] [CrossRef]
- Rutter, G.A. Dorothy Hodgkin Lecture 2014. Understanding genes identified by genome-wide association studies for type 2 diabetes. Diabet. Med. 2014, 31, 1480–1487. [Google Scholar] [CrossRef]
- Jonsson, A.; Ladenvall, C.; Ahluwalia, T.S.; Kravic, J.; Krus, U.; Taneera, J.; Isomaa, B.; Tuomi, T.; Renström, E.; Groop, L.; et al. Effects of Common Genetic Variants Associated with Type 2 Diabetes and Glycemic Traits on α- and β-Cell Function and Insulin Action in Humans. Diabetes 2013, 62, 2978–2983. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.-L.; Masiello, P.; Madiraju, S.M. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Hollingsworth, K.G.; Al-Mrabeh, A.; Avery, L.; Aribisala, B.; Caslake, M.; Taylor, R. Very Low-Calorie Diet and 6 Months of Weight Stability in Type 2 Diabetes: Pathophysiological Changes in Responders and Nonresponders. Diabetes Care 2016, 39, 808–815. [Google Scholar] [CrossRef]
- Taylor, R.; Al-Mrabeh, A.; Zhyzhneuskaya, S.; Peters, C.; Barnes, A.C.; Aribisala, B.S.; Hollingsworth, K.G.; Mathers, J.C.; Sattar, N.; Lean, M.E.J. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for beta cell recovery. Cell Metab. 2018, 28, 547–556.e3. [Google Scholar] [CrossRef]
- Taylor, R. Pathogenesis of type 2 diabetes: Tracing the reverse route from cure to cause. Diabetologia 2008, 51, 1781–1789. [Google Scholar] [CrossRef]
- Fabbrini, E.; Yoshino, J.; Yoshino, M.; Magkos, F.; Luecking, C.T.; Samovski, D.; Fraterrigo, G.; Okunade, A.L.; Patterson, B.W.; Klein, S. Metabolically normal obese people are protected from adverse effects following weight gain. J. Clin. Investig. 2015, 125, 787–795. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. β-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of Very Low–Density Lipoproteins Is the Hallmark of the Dyslipidemia in the Metabolic Syndrome. Arter. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered Lipid Metabolism and the Pathogenesis of Insulin Resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef]
- Fisher, E.A.; Cohen, D.E. Lipoprotein Metabolism, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2013, 33, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Poli, F.; Rodella, S.; Scala, L.; Tessari, R.; Zenari, L.; Falezza, G. Nonalcoholic Fatty Liver Disease and Risk of Future Cardiovascular Events Among Type 2 Diabetic Patients. Diabetes 2005, 54, 3541–3546. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A.; Peters, C.; Hollingsworth, K.G.; Taylor, R. Measurement of intraorgan fat and hepatic output of triglycerides in human type 2 diabetes by magnetic resonance and intralipid infusion techniques. STAR Protocols. In Press. [CrossRef]
- Al-Mrabeh, A.; Zhyzhneuskaya, S.V.; Peters, C.; Barnes, A.C.; Melhem, S.; Jesuthasan, A.; Aribisala, B.; Hollingsworth, K.G.; Lietz, G.; Mathers, J.C.; et al. Hepatic Lipoprotein Export and Remission of Human Type 2 Diabetes after Weight Loss. Cell Metab. 2020, 31, 233–249.e4. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 Diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Cline, G.W.; Wang, Y.; Rabin-Court, A.; Song, J.D.; Zhang, D.; Zhang, X.-M.; Nozaki, Y.; Dufour, S.; et al. Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. Cell Metab. 2018, 27, 210–217.e3. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-Induced CerS6-Dependent C16:0 Ceramide Production Promotes Weight Gain and Glucose Intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Ohman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty Acids, Obesity, and Insulin Resistance: Time for a Reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- Haas, M.E.; Attie, A.D.; Biddinger, S.B. The regulation of ApoB metabolism by insulin. Trends Endocrinol. Metab. 2013, 24, 391–397. [Google Scholar] [CrossRef]
- Sparks, J.D.; Dong, H.H. FoxO1 and hepatic lipid metabolism. Curr. Opin. Lipidol. 2009, 20, 217–226. [Google Scholar] [CrossRef]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, M. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Investig. 2006, 116, 2464–2472. [Google Scholar] [CrossRef]
- Åvall, K.; Ali, Y.; Leibiger, I.B.; Leibiger, B.; Moede, T.; Paschen, M.; Dicker, A.; Daré, E.; Köhler, M.; Ilegems, E.; et al. Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, E2611–E2619. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Bianconi, V.; Gragnano, F.; Moscarella, E.; Fimiani, F.; Monda, E.; Scudiero, O.; Limongelli, G.; Pirro, M.; Calabrò, P. Beyond cholesterol metabolism: The pleiotropic effects of proprotein convertase subtilisin/kexin type 9 (PCSK9). Genetics, mutations, expression, and perspective for long-term inhibition. BioFactors 2020, 46, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Norouzirad, R.; González-Muniesa, P.; Ghasemi, A. Hypoxia in Obesity and Diabetes: Potential Therapeutic Effects of Hyperoxia and Nitrate. Oxid. Med. Cell Longev. 2017, 2017, 5350267. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2019, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; Alonso, L.C. Lipotoxicity in the Pancreatic Beta Cell: Not Just Survival and Function, but Proliferation as Well? Curr. Diabetes Rep. 2014, 14, 492. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells during Development of Type 2 Diabetes. Front. Endocrinol. (Lausanne) 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Hauke, S.; Keutler, K.; Phapale, P.; Yushchenko, D.A.; Schultz, C. Endogenous Fatty Acids Are Essential Signaling Factors of Pancreatic β-Cells and Insulin Secretion. Diabetes 2018, 67, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V. Fatty Acids and Insulin Secretion: From FFAR and Near? Diabetes 2018, 67, 1932–1934. [Google Scholar] [CrossRef]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. β-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef]
- Pinnick, K.E.; Collins, S.C.; Londos, C.; Gauguier, D.; Clark, A.; Fielding, B.A. Pancreatic Ectopic Fat Is Characterized by Adipocyte Infiltration and Altered Lipid Composition. Obesity 2008, 16, 522–530. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.C.; Takahashi, H.K.; Dhayal, S.; Morgan, N.G.; Curi, R.; Newsholme, P. Arachidonic acid actions on functional integrity and attenuation of the negative effects of palmitic acid in a clonal pancreatic β-cell line. Clin. Sci. 2010, 120, 195–206. [Google Scholar] [CrossRef]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Green, C.D.; Olson, L.K. Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic β-cells by stearoyl-CoA desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E640–E649. [Google Scholar] [CrossRef]
- Zhao, H.; Matsuzaka, T.; Nakano, Y.; Motomura, K.; Tang, N.; Yokoo, T.; Okajima, Y.; Han, S.I.; Takeuchi, Y.; Aita, Y.; et al. Elovl6 Deficiency Improves Glycemic Control in Diabetic db/db Mice by Expanding β-Cell Mass and Increasing Insulin Secretory Capacity. Diabetes 2017, 66, 1833–1846. [Google Scholar] [CrossRef]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Fontés, G.; Zarrouki, B.; Hagman, D.K.; Latour, M.G.; Semache, M.; Roskens, V.; Moore, P.C.; Prentki, M.; Rhodes, C.J.; Jetton, T.L.; et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010, 53, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef]
- Kim-Muller, J.Y.; Zhao, S.; Srivastava, S.; Mugabo, Y.; Noh, H.L.; Kim, Y.R.; Madiraju, S.R.; Ferrante, A.W.; Skolnik, E.Y.; Prentki, M.; et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014, 20, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Kuok, I.T.; Rountree, A.M.; Jung, S.-R.; Sweet, I.R. Palmitate is not an effective fuel for pancreatic islets and amplifies insulin secretion independent of calcium release from endoplasmic reticulum. Islets 2019, 11, 51–64. [Google Scholar] [CrossRef]
- Rebelos, E.; Seghieri, M.; Natali, A.; Balkau, B.; Golay, A.; Piatti, P.M.; Lalic, N.M.; Laakso, M.; Mari, A.; Ferrannini, E. Influence of endogenous NEFA on beta cell function in humans. Diabetologia 2015, 58, 2344–2351. [Google Scholar] [CrossRef]
- Johnston, L.W.; Harris, S.B.; Retnakaran, R.; Giacca, A.; Liu, Z.; Bazinet, R.P.; Hanley, A.J. Association of NEFA composition with insulin sensitivity and beta cell function in the Prospective Metabolism and Islet Cell Evaluation (PROMISE) cohort. Diabetologia 2017, 61, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Salgin, B.; Ong, K.K.; Thankamony, A.; Emmett, P.; Wareham, N.J.; Dunger, D.B. Higher Fasting Plasma Free Fatty Acid Levels Are Associated with Lower Insulin Secretion in Children and Adults and a Higher Incidence of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Honka, H.; Hannukainen, J.C.; Tarkia, M.; Karlsson, H.; Saunavaara, V.; Salminen, P.; Soinio, M.; Mikkola, K.; Kudomi, N.; Oikonen, V.; et al. Pancreatic Metabolism, Blood Flow, and β-Cell Function in Obese Humans. J. Clin. Endocrinol. Metab. 2014, 99, E981–E990. [Google Scholar] [CrossRef]
- Tanabe, K.; Amo-Shiinoki, K.; Hatanaka, M.; Tanizawa, Y. Interorgan Crosstalk Contributing to β-Cell Dysfunction. J. Diabetes Res. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Pappan, K.L.; Pan, Z.; Kwon, G.; Marshall, C.A.; Coleman, T.; Goldberg, I.J.; McDaniel, M.L.; Semenkovich, C.F. Pancreatic beta-cell lipoprotein lipase independently regulates islet glucose metabolism and normal insulin secretion. J. Biol. Chem. 2005, 280, 9023–9029. [Google Scholar] [CrossRef]
- Gerst, F.; Wagner, R.; Kaiser, G.; Panse, M.; Heni, M.; Machann, J.; Bongers, M.N.; Sartorius, T.; Sipos, B.; Fend, F.; et al. Metabolic crosstalk between fatty pancreas and fatty liver: Effects on local inflammation and insulin secretion. Diabetologi 2017, 60, 2240–2251. [Google Scholar] [CrossRef]
- Tong, X.; Dai, C.; Walker, J.T.; Nair, G.G.; Kennedy, A.; Carr, R.M.; Hebrok, M.; Powers, A.C.; Stein, R. Lipid Droplet Accumulation in Human Pancreatic Islets Is Dependent On Both Donor Age and Health. Diabetes 2020, 69, 342–354. [Google Scholar] [CrossRef]
- Winzell, M.S.; Svensson, H.; Enerbäck, S.; Ravnskjaer, K.; Mandrup, S.; Esser, V.; Arner, P.; Alves-Guerra, M.C.; Miroux, B.; Sundler, F.; et al. Pancreatic beta-cell lipotoxicity induced by overexpression of hormone-sensitive lipase. Diabetes 2003, 52, 2057–2065. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54 (Suppl. S2), S108–S113. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T.; et al. β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition. Am. J. Pathol. 2011, 178, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet β-Cells in Type 2 Diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- White, M.G.; Marshall, H.L.; Rigby, R.; Huang, G.C.; Amer, A.; Booth, T.; White, S.; Shaw, J.A. Expression of mesenchymal and alpha-cell phenotypic markers in islet beta-cells in recently diagnosed diabetes. Diabetes Care 2013, 36, 3818–3820. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Anderson, S.J.; White, M.G.; Armour, S.L.; Maheshwari, R.; Tiniakos, D.; Muller, Y.D.; Berishvili, E.; Berney, T.; Shaw, J.A.M. Loss of end-differentiated β-cell phenotype following pancreatic islet transplantation. Arab. Archaeol. Epigr. 2017, 18, 750–755. [Google Scholar] [CrossRef]
- White, M.G.; Shaw, J.A.M.; Taylor, R. Type 2 diabetes: The pathologic basis of reversible beta-cell dysfunction. Diabetes Care 2016, 39, 2080–2088. [Google Scholar] [CrossRef]
- Brereton, M.F.; Iberl, M.; Shimomura, K.; Zhang, Q.; Adriaenssens, A.E.; Proks, P.; Spiliotis, I.I.; Dace, W.; Mattis, K.K.; Ramracheya, R.; et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat. Commun. 2014, 5, 4639. [Google Scholar] [CrossRef]
- Cnop, M.; Grupping, A.; Hoorens, A.; Bouwens, L.; Pipeleers-Marichal, M.; Pipeleers, D. Endocytosis of low-density lipoprotein by human pancreatic beta cells and uptake in lipid-storing vesicles, which increase with age. Am. J. Pathol. 2000, 156, 237–244. [Google Scholar] [CrossRef]
- Roehrich, M.E.; Mooser, V.; Lenain, V.; Herz, J.; Nimpf, J.; Azhar, S.; Bideau, M.; Capponi, A.; Nicod, P.; Haefliger, J.A.; et al. Insulin-secreting beta-cell dysfunction induced by human lipoproteins. J. Biol. Chem. 2003, 278, 18368–18375. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Kerksiek, A.; Gebert, K.; Annema, W.; Sibler, R.; Radosavljevic, S.; Luetjohann, D.; Rohrer, L.; Von Eckardstein, A. HDL inhibits endoplasmic reticulum stress-induced apoptosis of pancreatic β-cells in vitro by activation of Smoothened. J. Lipid Res. 2020, 61, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Steven, S.; Tiniakos, D.; Taylor, R. Quantification of intrapancreatic fat in type 2 diabetes by MRI. PLoS ONE 2017, 12, e0174660. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Suleiman, M.; Marselli, L. Organ donor pancreases for the study of human islet cell histology and pathophysiology: A precious and valuable resource. Diabetologia 2018, 61, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.-Y.; Soetedjo, A.A.P.; Amirruddin, N.S.; Chang, Y.-T.; Eriksson, O.; Teo, A.K.K. Tools for Bioimaging Pancreatic β Cells in Diabetes. Trends Mol. Med. 2019, 25, 708–722. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pipeleers, D.G.; Ling, Z.; Welsh, N.; Hellerstrom, C.; Andersson, A. Major species differences between humans and rodents in the susceptibility to pancreatic beta-cell injury. Proc. Natl. Acad. Sci. USA 1994, 91, 9253–9256. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.J.; Kim, A.; Miller, K.; Hara, M. Pancreatic islet plasticity: Interspecies comparison of islet architecture and composition. Islets 2010, 2, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S.; Li, W.C.; Ouziel-Yahalom, L.; Guo, L.; Weir, G.C.; Sharma, A. Beta-cell growth and regeneration: Replication is only part of the story. Diabetes 2010, 59, 2340–2348. [Google Scholar] [CrossRef]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Da Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.-V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA Sequencing Identifies Dysregulation of the Human Pancreatic Islet Transcriptome by the Saturated Fatty Acid Palmitate. Diabetes 2013, 63, 1978–1993. [Google Scholar] [CrossRef]
- Nagao, M.; Esguerra, J.L.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in β-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction. In Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2020. [Google Scholar]
- Havel, P.J.; Kasim-Karakas, S.; Dubuc, G.R.; Muller, W.; Phinney, S.D. Gender differences in plasma leptin concentrations. Nat. Med. 1996, 2, 949–950. [Google Scholar] [CrossRef]
- Ohman-Hanson, R.A.; Cree-Green, M.; Kelsey, M.M.; Bessesen, D.H.; Sharp, T.A.; Pyle, L.; Pereira, R.I.; Nadeau, K.J. Ethnic and Sex Differences in Adiponectin: From Childhood to Adulthood. J. Clin. Endocrinol. Metab. 2016, 101, 4808–4815. [Google Scholar] [CrossRef]
- Lekva, T.; Michelsen, A.E.; Aukrust, P.; Henriksen, T.; Bollerslev, J.; Ueland, T. Leptin and adiponectin as predictors of cardiovascular risk after gestational diabetes mellitus. Cardiovasc. Diabetol. 2017, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Naruse, M.; Usui, T.; Tagami, T.; Suganami, T.; Yamada, K.; Kuzuya, H.; Shimatsu, A.; Ogawa, Y. Leptin-to-adiponectin ratio as a potential atherogenic index in obese type 2 diabetic patients. Diabetes Care 2004, 27, 2488–2490. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less Is More. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Long-Term Persistence of Hormonal Adaptations to Weight Loss. N. Engl. J. Med. 2011, 365, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T.; Roth, C.; Menke, T.; Andler, W. Adiponectin before and after Weight Loss in Obese Children. J. Clin. Endocrinol. Metab. 2004, 89, 3790–3794. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, W.; Huang, T.; Zheng, Y.; Wang, M.; Bray, G.A.; Sacks, F.M.; Qi, L. Weight-Loss Diets, Adiponectin, and Changes in Cardiometabolic Risk in the 2-Year POUNDS Lost Trial. J. Clin. Endocrinol. Metab. 2016, 101, 2415–2422. [Google Scholar] [CrossRef]
- Nicklas, B.J.; Katzel, L.I.; Ryan, A.S.; Dennis, K.E.; Goldberg, A.P. Gender Differences in the Response of Plasma Leptin Concentrations to Weight Loss in Obese Older Individuals. Obes. Res. 1997, 5, 62–68. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. Rev. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- Poreba, M.; Rostoff, P.; Siniarski, A.; Mostowik, M.; Golebiowska-Wiatrak, R.; Nessler, J.; Undas, A.; Gajos, G. Relationship between polyunsaturated fatty acid composition in serum phospholipids, systemic low-grade inflammation, and glycemic control in patients with type 2 diabetes and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 29. [Google Scholar] [CrossRef]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B. Interplay between Lipids and Branched-Chain Amino Acids in Development of Insulin Resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Beltran, V.D.; Chan, S.M.; Brown, J.R.; Bevington, A.; Herbert, T.P. System-L amino acid transporters play a key role in pancreatic beta-cell signalling and function. J. Mol. Endocrinol. 2016, 56, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Mattson, M.P.; Allison, D.B.; Fontana, L.; Harvie, M.; Longo, V.D.; Malaisse, W.J.; Mosley, M.; Notterpek, L.; Ravussin, E.; Scheer, F.A.J.L.; et al. Meal frequency and timing in health and disease. Proc. Natl. Acad. Sci. USA 2014, 111, 16647–16653. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular Mechanisms and Clinical Applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.H.; Crosslin, D.R.; Haynes, C.S.; Nelson, S.; Turer, C.B.; Stevens, R.D.; Muehlbauer, M.J.; Wenner, B.R.; Bain, J.R.; Laferrère, B.; et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetol. 2011, 55, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Lips, M.A.; Van Klinken, J.B.; Van Harmelen, V.; Dharuri, H.K.; Hoen, P.A.T.; Laros, J.F.; Van Ommen, G.-J.; Janssen, I.M.; Van Ramshorst, B.; Van Wagensveld, B.A.; et al. Roux-en-Y Gastric Bypass Surgery, but Not Calorie Restriction, Reduces Plasma Branched-Chain Amino Acids in Obese Women Independent of Weight Loss or the Presence of Type 2 Diabetes. Diabetes Care 2014, 37, 3150–3156. [Google Scholar] [CrossRef]
- Patel, S.; Alvarez-Guaita, A.; Melvin, A.; Rimmington, D.; Dattilo, A.; Miedzybrodzka, E.L.; Cimino, I.; Maurin, A.-C.; Roberts, G.P.; Meek, C.L.; et al. GDF15 Provides an Endocrine Signal of Nutritional Stress in Mice and Humans. Cell Metab. 2019, 29, 707–718.e8. [Google Scholar] [CrossRef]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.-S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.-B.; et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity Is a Fibroblast Growth Factor 21 (FGF21)-Resistant State. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef]
- Chen, W.; Hoo, R.L.-C.; Konishi, M.; Itoh, N.; Lee, P.-C.; Ye, H.-Y.; Lam, K.S.-L.; Xu, A. Growth Hormone Induces Hepatic Production of Fibroblast Growth Factor 21 through a Mechanism Dependent on Lipolysis in Adipocytes*. J. Biol. Chem. 2011, 286, 34559–34566. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Van De Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef]
- Chen, C.; Cheung, B.M.; Tso, A.W.; Wang, Y.; Law, L.S.; Ong, K.L.; Wat, N.M.; Xu, A.; Lam, K.S. High Plasma Level of Fibroblast Growth Factor 21 Is an Independent Predictor of Type 2 Diabetes: A 5.4-year population-based prospective study in Chinese subjects. Diabetes Care 2011, 34, 2113–2115. [Google Scholar] [CrossRef]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-Adiponectin-Ceramide Axis Controls Energy Expenditure and Insulin Action in Mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Scherer, P.E. Lowering ceramides to overcome diabetes. Science 2019, 365, 319–320. [Google Scholar] [CrossRef]
- Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, S.H.; Kim, S.-N.; Kwon, H.-J.; Kim, J.-D.; Oh, J.Y.; Jung, Y.-S. Sex-specific metabolic interactions between liver and adipose tissue in MCD diet-induced non-alcoholic fatty liver disease. Oncotarget 2016, 7, 46959–46971. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Schlein, C.; Talukdar, S.; Heine, M.; Fischer, A.W.; Krott, L.M.; Nilsson, S.K.; Brenner, M.B.; Heeren, J.; Scheja, L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016, 23, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.C.; Turner, S.M.; Thomas, K.S.A.; McCrudden, P.R.; Fine, D.R.; Johnson, P.A.; Johnson, C.D.; Iredale, J.P. Apoptosis and proliferation of acinar and islet cells in chronic pancreatitis: Evidence for differential cell loss mediating preservation of islet function. Gut 2002, 50, 542–548. [Google Scholar] [CrossRef]
- Das, S.L.M.; Kennedy, J.I.C.; Murphy, R.; Phillips, A.R.J.; Windsor, J.A.; Petrov, M.S. Relationship between the exocrine and endocrine pancreas after acute pancreatitis. World J. Gastroenterol. 2014, 20, 17196–17205. [Google Scholar] [CrossRef]
- Czako, L.; Hegyi, P.; Rakonczay, Z.; Wittmann, T.; Otsuki, M. Interactions between the Endocrine and Exocrine Pancreas and Their Clinical Relevance. Pancreatology 2009, 9, 351–359. [Google Scholar] [CrossRef]
- Piciucchi, M.; Capurso, G.; Archibugi, L.; Fave, M.M.D.; Capasso, M.; Fave, G.D. Exocrine Pancreatic Insufficiency in Diabetic Patients: Prevalence, Mechanisms, and Treatment. Int. J. Endocrinol. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Steven, S.; Taylor, R. Morphology of the pancreas in type 2 diabetes: Effect of weight loss with or without normalisation of insulin secretory capacity. Diabetologia 2016, 59, 1753–1759. [Google Scholar] [CrossRef]
- Macauley, M.; Percival, K.; Thelwall, P.E.; Hollingsworth, K.G.; Taylor, R. Altered Volume, Morphology and Composition of the Pancreas in Type 2 Diabetes. PLoS ONE 2015, 10, e0126825. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Hollingsworth, K.G.; Small, P.K.; Woodcock, S.A.; Pucci, A.; Aribisala, B.; Al-Mrabeh, A.; Daly, A.K.; Batterham, R.L.; Taylor, R. Weight Loss Decreases Excess Pancreatic Triacylglycerol Specifically in Type 2 Diabetes. Diabetes Care 2015, 39, 158–165. [Google Scholar] [CrossRef]
- Zhyzhneuskaya, S.V.; Al-Mrabeh, A.; Peters, C.; Barnes, A.; Aribisala, B.; Hollingsworth, K.G.; McConnachie, A.; Sattar, N.; Lean, M.E.; Taylor, R. Time Course of Normalization of Functional β-Cell Capacity in the Diabetes Remission Clinical Trial After Weight Loss in Type 2 Diabetes. Diabetes Care 2020, 43, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. (Edinb. Scotl.) 1988, 9, 151–159. [Google Scholar]
- Matsuda, A.; Makino, N.; Tozawa, T.; Shirahata, N.; Honda, T.; Ikeda, Y.; Sato, H.; Ito, M.; Kakizaki, Y.; Akamatsu, M.; et al. Pancreatic Fat Accumulation, Fibrosis, and Acinar Cell Injury in the Zucker Diabetic Fatty Rat Fed a Chronic High-Fat Diet. Pancreas 2014, 43, 735–743. [Google Scholar] [CrossRef]
- Moran, A.; Brunzell, C.; Cohen, R.C.; Katz, M.; Marshall, B.C.; Onady, G.; Robinson, K.A.; Sabadosa, K.A.; Stecenko, A.; Slovis, B.; et al. Clinical Care Guidelines for Cystic Fibrosis-Related Diabetes: A position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 2010, 33, 2697–2708. [Google Scholar] [CrossRef]
- Hart, N.J.; Aramandla, R.; Poffenberger, G.; Fayolle, C.; Thames, A.H.; Bautista, A.; Spigelman, A.F.; Babon, J.A.B.; DeNicola, M.E.; Dadi, P.K.; et al. Cystic fibrosis–related diabetes is caused by islet loss and inflammation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Bonal, C.; Thorel, F.; Ait-Lounis, A.; Reith, W.; Trumpp, A.; Herrera, P.L. Pancreatic Inactivation of c-Myc Decreases Acinar Mass and Transdifferentiates Acinar Cells into Adipocytes in Mice. Gastroenterology 2009, 136, 309–319.e9. [Google Scholar] [CrossRef] [PubMed]
- Leroith, D.; Yakar, S. Mechanisms of Disease: Metabolic effects of growth hormone and insulin-like growth factor 1. Nat. Clin. Pr. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Goda, K.; Sasaki, E.; Nagata, K.; Fukai, M.; Ohsawa, N.; Hahafusa, T. Pancreatic volume in type 1 und type 2 diabetes mellitus. Acta Diabetol. 2001, 38, 145–149. [Google Scholar] [CrossRef]
- Williams, A.J.K.; Thrower, S.L.; Sequeiros, I.M.; Ward, A.; Bickerton, A.S.; Triay, J.M.; Callaway, M.P.; Dayan, C.M. Pancreatic Volume Is Reduced in Adult Patients with Recently Diagnosed Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 2109–2113. [Google Scholar] [CrossRef]
- Carey, P.E.; Halliday, J.; Snaar, J.E.M.; Morris, P.G.; Taylor, R. Direct assessment of muscle glycogen storage after mixed meals in normal and type 2 diabetic subjects. Am. J. Physiol. Metab. 2003, 284, E688–E694. [Google Scholar] [CrossRef]
- Carroll, P.V.; Christ, E.R.; Umpleby, A.M.; Gowrie, I.; Jackson, N.; Bowes, S.B.; Hovorka, R.; Croos, P.; Sonksen, P.H.; Russell-Jones, D.L. IGF-I treatment in adults with type 1 diabetes: Effects on glucose and protein metabolism in the fasting state and during a hyperinsulinemic-euglycemic amino acid clamp. Diabetes 2000, 49, 789–796. [Google Scholar] [CrossRef]
- Friedrich, N.; Thuesen, B.; Jørgensen, T.; Juul, A.; Spielhagen, C.; Wallaschofksi, H.; Linneberg, A. The Association Between IGF-I and Insulin Resistance: A general population study in Danish adults. Diabetes Care 2012, 35, 768–773. [Google Scholar] [CrossRef]
- Cohen, D.H.; Leroith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Milman, S.; Huffman, D.M.; Barzilai, N. The Somatotropic Axis in Human Aging: Framework for the Current State of Knowledge and Future Research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Shaw, J.A.M.; McConnachie, A.; Sattar, N.; Lean, M.E.J.; Taylor, R. 2-year remission of type 2 diabetes and pancreas morphology: A post-hoc analysis of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2020, 8, 939–948. [Google Scholar] [CrossRef]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nat. Cell Biol. 2018, 557, 351–358. [Google Scholar] [CrossRef]
- Dokken, B.B. The Pathophysiology of Cardiovascular Disease and Diabetes: Beyond Blood Pressure and Lipids. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar] [CrossRef]
- Krauss, R.M. Lipids and Lipoproteins in Patients with Type 2 Diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- LaMantia, V.; Sniderman, A.; Faraj, M. Nutritional management of hyperapoB. Nutr. Res. Rev. 2016, 29, 202–233. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.F.; Benseñor, I.M.; Nogueira, A.C.C.; Duncan, B.B.; Schmidt, M.I.; Blaha, M.J.; Toth, P.P.; Jones, S.R.; Santos, R.D.; Lotufo, P.A.; et al. Increased particle size of triacylglycerol-enriched remnant lipoproteins, but not their plasma concentration or lipid content, augments risk prediction of incident type 2 diabetes. Diabetologia 2021, 64, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Alkhalil, M.; Andreotti, F.; Maggioni, A.P.; Ferro, C.J.; Sarafidis, P.; Ortiz, A.; Tsimikas, S.; Karwatowska-Prokopczuk, E.; Xia, S. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, e65. [Google Scholar] [CrossRef]
- Welsh, P.; Welsh, C.; Celis-Morales, C.; Brown, R.; Ferguson, L.; Gray, S.; Mark, P.; Lewsey, J.; Lyall, D.; Gill, J.; et al. Lipoprotein(a) and cardiovascular disease: Prediction, attributable risk fraction and estimating benefits from novel interventions. Eur. Heart J. 2020, 41. [Google Scholar] [CrossRef]
- Knouff, C.; Hinsdale, M.E.; Mezdour, H.; Altenburg, M.K.; Watanabe, M.; Quarfordt, S.H.; Sullivan, P.M.; Maeda, N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Investig. 1999, 103, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, O.; Martinelli, N.; Bassi, A.; Trabetti, E.; Girelli, D.; Pizzolo, F.; Friso, S.; Pignatti, P.F.; Corrocher, R. ApoE epsilon2/epsilon3/epsilon4 polymorphism, ApoC-III/ApoE ratio and metabolic syndrome. Clin. Exp. Med. 2007, 7, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Pechlaner, R.; Tsimikas, S.; Yin, X.; Willeit, P.; Baig, F.; Santer, P.; Oberhollenzer, F.; Egger, G.; Witztum, J.L.; Alexander, V.J.; et al. Very-Low-Density Lipoprotein–Associated Apolipoproteins Predict Cardiovascular Events and Are Lowered by Inhibition of APOC-III. J. Am. Coll. Cardiol. 2017, 69, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Gordts, P.L.; Nock, R.; Son, N.H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef]
- Ramms, B.; Patel, S.; Nora, C.; Pessentheiner, A.R.; Chang, M.W.; Green, C.R.; Golden, G.J.; Secrest, P.; Krauss, R.M.; Metallo, C.M.; et al. ApoC-III ASO promotes tissue LPL activity in the absence of apoE-mediated TRL clearance. J. Lipid Res. 2019, 60, 1379–1395. [Google Scholar] [CrossRef] [PubMed]
- Mooijaart, S.P.; Berbée, J.F.P.; Van Heemst, D.; Havekes, L.M.; De Craen, A.J.M.; Slagboom, P.E.; Rensen, P.C.N.; Westendorp, R.G.J. ApoE Plasma Levels and Risk of Cardiovascular Mortality in Old Age. PLoS Med. 2006, 3, e176. [Google Scholar] [CrossRef] [PubMed]
- Sofat, R.; Cooper, J.A.; Kumari, M.; Casas, J.P.; Mitchell, J.P.; Acharya, J.; Thom, S.; Hughes, A.D.; Humphries, S.E.; Hingorani, A.D. Circulating Apolipoprotein E Concentration and Cardiovascular Disease Risk: Meta-analysis of Results from Three Studies. PLoS Med. 2016, 13, e1002146. [Google Scholar] [CrossRef]
- Atzmon, G.; Rincon, M.; Schechter, C.B.; Shuldiner, A.R.; Lipton, R.B.; Bergman, A.; Barzilai, N. Lipoprotein Genotype and Conserved Pathway for Exceptional Longevity in Humans. PLoS Biol. 2006, 4, e113. [Google Scholar] [CrossRef]
- Huebschmann, A.G.; Huxley, R.R.; Kohrt, W.M.; Zeitler, P.; Regensteiner, J.G.; Reusch, J.E.B. Sex differences in the burden of type 2 diabetes and cardiovascular risk across the life course. Diabetologia 2019, 62, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef] [PubMed]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J. Clin. Endocrinol. Metab. 2015, 100, 4425–4433. [Google Scholar] [CrossRef]
- Grant, P.J.; Cosentino, F. The 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: New features and the ‘Ten Commandments’ of the 2019 Guidelines are discussed by Professor Peter J. Grant and Professor Francesco Cosentino, the Task Force chairmen. Eur. Heart J. 2019, 40, 3215–3217. [Google Scholar] [CrossRef]
- Taylor, R. Type 2 Diabetes: Etiology and reversibility. Diabetes Care 2013, 36, 1047–1055. [Google Scholar] [CrossRef]
- Thom, G.; McIntosh, A.; Messow, C.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; McCombie, L.; Malkova, D.; Al-Mrabeh, A.; Zhyzhneuskaya, S.; et al. Weight loss-induced increase in fasting ghrelin concentration is a predictor of weight regain: Evidence from the Diabetes Remission Clinical Trial (DiRECT). Diabetes Obes. Metab. 2020, 23, 711–719. [Google Scholar] [CrossRef]
- Mai, K.; Li, L.; Wiegand, S.; Brachs, M.; Leupelt, V.; Ernert, A.; Kühnen, P.; Hübner, N.; Robinson, P.; Chen, W.; et al. An Integrated Understanding of the Molecular Mechanisms of How Adipose Tissue Metabolism Affects Long-term Body Weight Maintenance. Diabetes 2019, 68, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Batterham, R.L.; Cummings, D.E. Mechanisms of Diabetes Improvement Following Bariatric/Metabolic Surgery. Diabetes Care 2016, 39, 893–901. [Google Scholar] [CrossRef]
- Lingvay, I.; Guth, E.; Islam, A.; Livingston, E. Rapid improvement in diabetes after gastric bypass surgery: Is it the diet or surgery? Diabetes Care 2013, 36, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Kayser, B.D.; Yoshino, J.; Stein, R.I.; Reeds, D.; Eagon, J.C.; Eckhouse, S.R.; Watrous, J.D.; Jain, M.; Knight, R.; et al. Effects of Diet versus Gastric Bypass on Metabolic Function in Diabetes. N. Engl. J. Med. 2020, 383, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Martinussen, C.; Bojsen-Møller, K.N.; Dirksen, C.; Jacobsen, S.H.; Jørgensen, N.B.; Kristiansen, V.B.; Holst, J.J.; Madsbad, S. Immediate enhancement of first-phase insulin secretion and unchanged glucose effectiveness in patients with type 2 diabetes after Roux-en-Y gastric bypass. Am. J. Physiol. Metab. 2015, 308, E535–E544. [Google Scholar] [CrossRef]
- Steven, S.; Hollingsworth, K.G.; Small, P.K.; Woodcock, S.A.; Pucci, A.; Aribasala, B.; Al-Mrabeh, A.; Batterham, R.L.; Taylor, R. Calorie restriction and not glucagon-like peptide-1 explains the acute improvement in glucose control after gastric bypass in Type 2 diabetes. Diabet. Med. 2016, 33, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef]
- Karakose, E.; Ackeifi, C.; Wang, P.; Stewart, A.F. Advances in drug discovery for human beta cell regeneration. Diabetologia 2018, 61, 1693–1699. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e555. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [PubMed]
- Castano-Martinez, T.; Schumacher, F.; Schumacher, S.; Kochlik, B.; Weber, D.; Grune, T.; Biemann, R.; McCann, A.; Abraham, K.; Weikert, C.; et al. Methionine restriction prevents onset of type 2 diabetes in NZO mice. Faseb J. 2019, 33, 7092–7102. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef]
- Muyyarikkandy, M.S.; McLeod, M.; Maguire, M.; Mahar, R.; Kattapuram, N.; Zhang, C.; Surugihalli, C.; Muralidaran, V.; Vavilikolanu, K.; Mathews, C.E.; et al. Branched chain amino acids and carbohydrate restriction exacerbate ketogenesis and hepatic mitochondrial oxidative dysfunction during NAFLD. Faseb J. 2020, 34, 14832–14849. [Google Scholar] [CrossRef]
- Van Zuuren, E.J.; Fedorowicz, Z.; Kuijpers, T.; Pijl, H. Effects of low-carbohydrate- compared with low-fat-diet interventions on metabolic control in people with type 2 diabetes: A systematic review including GRADE assessments. Am. J. Clin. Nutr. 2018, 108, 300–331. [Google Scholar] [CrossRef]
- Inaishi, J.; Saisho, Y. Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review. Nutrients 2020, 12, 3846. [Google Scholar] [CrossRef]
- Yaney, G.C.; Corkey, B.E. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 2003, 46, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Curletto, G.; Cipullo, D.; Rigault de la Longrais, R.; Trento, M.; Passera, P.; Taulaigo, A.V.; Di Miceli, S.; Cenci, A.; Dalmasso, P.; et al. Estimating the delay between onset and diagnosis of type 2 diabetes from the time course of retinopathy prevalence. Diabetes Care 2014, 37, 1668–1674. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Weitz, J.; Panzer, J.K.; Klein, D.; Moreno-Hernández, Y.; Cechin, S.; Tamayo, A.; Almaça, J.; Hiller, H.; et al. Long-term culture of human pancreatic slices as a model to study real-time islet regeneration. Nat. Commun. 2020, 11, 3265. [Google Scholar] [CrossRef]
- Debois, D.; Bralet, M.P.; Le Naour, F.; Brunelle, A.; Laprevote, O. In situ lipidomic analysis of nonalcoholic fatty liver by cluster TOF-SIMS imaging. Anal. Chem. 2009, 81, 2823–2831. [Google Scholar] [CrossRef]
- Passarelli, M.K.; Winograd, N. Lipid imaging with time-of-flight secondary ion mass spectrometry (ToF-SIMS). Biochim Biophys Acta 2011, 1811, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Prentice, B.M.; Hart, N.J.; Phillips, N.; Haliyur, R.; Judd, A.; Armandala, R.; Spraggins, J.M.; Lowe, C.L.; Boyd, K.L.; Stein, R.W.; et al. Imaging mass spectrometry enables molecular profiling of mouse and human pancreatic tissue. Diabetologia 2019, 62, 1036–1047. [Google Scholar] [CrossRef]
- Wang, Y.J.; Traum, D.; Schug, J.; Gao, L.; Liu, C.; Atkinson, M.A.; Powers, A.C.; Feldman, M.D.; Naji, A.; Chang, K.M.; et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019, 29, 769–783.e764. [Google Scholar] [CrossRef] [PubMed]
- Damond, N.; Engler, S.; Zanotelli, V.R.T.; Schapiro, D.; Wasserfall, C.H.; Kusmartseva, I.; Nick, H.S.; Thorel, F.; Herrera, P.L.; Atkinson, M.A.; et al. A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab. 2019, 29, 755–768.e755. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Minami, K.; Okuno, M.; Miyawaki, K.; Okumachi, A.; Ishizaki, K.; Oyama, K.; Kawaguchi, M.; Ishizuka, N.; Iwanaga, T.; Seino, S. Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 2005, 102, 15116–15121. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef]
- Ravnskjaer, K.; Boergesen, M.; Rubi, B.; Larsen, J.K.; Nielsen, T.; Fridriksson, J.; Maechler, P.; Mandrup, S. Peroxisome proliferator-activated receptor alpha (PPARalpha) potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology 2005, 146, 3266–3276. [Google Scholar] [CrossRef]
- Trevino, M.B.; Machida, Y.; Hallinger, D.R.; Garcia, E.; Christensen, A.; Dutta, S.; Peake, D.A.; Ikeda, Y.; Imai, Y. Perilipin 5 regulates islet lipid metabolism and insulin secretion in a cAMP-dependent manner: Implication of its role in the postprandial insulin secretion. Diabetes 2015, 64, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, X.; Zhang, L.; Zhang, M.; Li, L.; Luo, D.; Zhong, Y. Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic β-cells. Nutr. Metab. (Lond.) 2019, 16, 50. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).