
Oligonucleotide-Based Therapies for Renal Diseases

 , , ,
, , , 
Abstract
:1. Introduction
2. Oligonucleotides Used in Therapeutics
2.1. RNA-Based Strategies
2.1.1. siRNA
2.1.2. saRNA
2.1.3. miRNA
2.2. RNA/Protein-Based Strategies (CRISPR)
2.3. DNA/RNA-Based Strategies
2.3.1. Antisense Oligonucleotides (ASOs)
2.3.2. Aptamers
2.4. DNA-Based Strategies
Transcription Factor Decoy (TFD)
3. Overcoming Delivery Problems
3.1. Chemical Modifications to Improve Stability and Biodistribution
3.2. Improving Kidney Delivery
3.2.1. In Vivo Local Delivery
3.2.2. Viral Delivery
3.2.3. Nanocarriers
3.2.4. Nanoparticles
3.2.5. Aptamers
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Li, P.K.T.; Garcia-Garcia, G.; Lui, S.F.; Andreoli, S.; Fung, W.W.S.; Hradsky, A.; Kumaraswami, L.; Liakopoulos, V.; Rakhimova, Z.; Saadi, G.; et al. Kidney Health for Everyone Everywhere: From Prevention to Detection and Equitable Access to Care. Am. J. Hypertens 2020, 33, 282–289. [Google Scholar] [CrossRef]
- Devuyst, O.; Knoers, N.V.A.M.; Remuzzi, G.; Schaefer, F. Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014, 383, 1844–1859. [Google Scholar] [CrossRef] [Green Version]
- Augustine, J. Kidney transplant: New opportunities and challenges. Cleve Clin. J. Med. 2018, 85, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- McCaffrey, A.P. RNA interference inhibitors of hepatitis B virus. In Proceedings of the Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Hoboken, NJ, USA, 2009; Volume 1175, pp. 15–23. [Google Scholar]
- Jackson, A.L.; Linsley, P.S. Noise amidst the silence: Off-target effects of siRNAs? Trends Genet. 2004, 20, 521–524. [Google Scholar] [CrossRef]
- Anderson, E.; Boese, Q.; Khvorova, A.; Karpilow, J. Identifying siRNA-induced off-targets by microarray analysis. Methods Mol. Biol. 2008, 442, 45–63. [Google Scholar] [CrossRef]
- Yoshida, T.; Naito, Y.; Yasuhara, H.; Sasaki, K.; Kawaji, H.; Kawai, J.; Naito, M.; Okuda, H.; Obika, S.; Inoue, T. Evaluation of off-target effects of gapmer antisense oligonucleotides using human cells. Genes Cells 2019, 24, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Edvard Smith, C.I.; Zain, R. Therapeutic oligonucleotides: State of the art. Annu. Rev. Pharmacol. Toxicol. 2018, 59, 605–630. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, X. Aptamer-based targeted therapy. Adv. Drug Deliv. Rev. 2018, 134, 65–78. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molitoris, B.A.; Dagher, P.C.; Sandoval, R.M.; Campos, S.B.; Ashush, H.; Fridman, E.; Brafman, A.; Faerman, A.; Atkinson, S.J.; Thompson, J.D.; et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Takabatake, Y.; Isaka, Y.; Imai, E. In vivo Transfer of Small Interfering RNA or Small Hairpin RNA Targeting Glomeruli. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2009; Volume 466, pp. 251–263. [Google Scholar]
- Shimizu, H.; Hori, Y.; Kaname, S.; Yamada, K.; Nishiyama, N.; Matsumoto, S.; Miyata, K.; Oba, M.; Yamada, A.; Kataoka, K.; et al. SiRNA-based therapy ameliorates glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Hein, S.; Dagnæs-Hansen, F.; Weyer, K.; Yang, C.; Nielsen, R.; Christensen, E.I.; Fenton, R.A.; Kjems, J. Megalin-mediated specific uptake of chitosan/siRNA nanoparticles in mouse kidney proximal tubule epithelial cells enables AQP1 gene silencing. Theranostics 2014, 4, 1039–1051. [Google Scholar] [CrossRef] [Green Version]
- Morishita, Y.; Yoshizawa, H.; Watanabe, M.; Ishibashi, K.; Muto, S.; Kusano, E.; Nagata, D. SiRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Nilsson, L.; Cheema, M.U.; Wang, Y.; Frøkiær, J.; Gao, S.; Kjems, J.; Nørregaard, R. Chitosan/siRNA nanoparticles targeting cyclooxygenase type 2 attenuate unilateral ureteral obstruction-induced kidney injury in mice. Theranostics 2015, 5, 110–123. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Gale, A.; Wu, P.; Ma, R.; Davis, M.E. SiRNA delivery to the glomerular mesangium using polycationic cyclodextrin nanoparticles containing siRNA. Nucleic Acid Ther. 2015, 25, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zang, G.Y.; Jiang, J.; He, W.; Johnston, N.J.; Ling, H.; Chen, R.; Zhang, X.; Liu, Y.; Haig, A.; et al. Attenuating ischemia-reperfusion injury in kidney transplantation by perfusing donor organs with siRNA cocktail solution. Transplantation 2016, 100, 743–752. [Google Scholar] [CrossRef]
- Alidori, S.; Akhavein, N.; Thorek, D.L.J.; Behling, K.; Romin, Y.; Queen, D.; Beattie, B.J.; Manova-Todorova, K.; Bergkvist, M.; Scheinberg, D.A.; et al. Targeted fibrillar nanocarbon RNAi treatment of acute kidney injury. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Eadon, M.T.; Cheng, Y.-H.; Hato, T.; Benson, E.A.; Ipe, J.; Collins, K.S.; De Luca, T.; El-Achkar, T.M.; Bacallao, R.L.; Skaar, T.C.; et al. In vivo siRNA Delivery and Rebound of Renal LRP2 in Mice. J. Drug Deliv. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Narváez, A.; Guiteras, R.; Sola, A.; Manonelles, A.; Morote, J.; Torras, J.; Grinyó, J.M.; Cruzado, J.M. SiRNA-silencing of CD40 attenuates unilateral ureteral obstruction-induced kidney injury in mice. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Q.; Wang, J.; Li, L.; Sun, X.; Zhang, Z.; Zhang, L. Co-delivery of p38α MAPK and p65 siRNA by novel liposomal glomerulus-targeting nano carriers for effective immunoglobulin a nephropathy treatment. J. Control. Release 2020, 320, 457–468. [Google Scholar] [CrossRef]
- Thai, H.B.D.; Kim, K.R.; Hong, K.T.; Voitsitskyi, T.; Lee, J.S.; Mao, C.; Ahn, D.R. Kidney-Targeted Cytosolic Delivery of siRNA Using a Small-Sized Mirror DNA Tetrahedron for Enhanced Potency. ACS Cent. Sci. 2020. [Google Scholar] [CrossRef]
- Wang, X.; Skelley, L.; Cade, R.; Sun, Z. AAV delivery of mineralocorticoid receptor shRNA prevents progression of cold-induced hypertension and attenuates renal damage. Gene Ther. 2006, 13, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Shou, Z.; Xiao, H.; Xu, Y.; Wang, Y.; Yang, Y.; Jiang, H.; Chen, J.; Yamada, K.; Miyamoto, K. SHARP-2 gene silencing by lentiviral-based short hairpin RNA interference prolonged rat kidney transplant recipients’ survival time. J. Int. Med. Res. 2009, 37, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Shan, Y.; Zhao, H.; He, P. Biological effects of lentivirus-mediated shRNA targeting collagen type i on the mesangial cells of rats. Ren. Fail. 2011, 33, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, T.; Muhib, S.; Sato, N.; Hasebe, N. Silencing of p53 RNA through transarterial delivery ameliorates renal tubular injury and downregulates GSK-3β expression after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2013, 305. [Google Scholar] [CrossRef] [PubMed]
- Espana-Agusti, J.; Tuveson, D.A.; Adams, D.J.; Matakidou, A. A minimally invasive, lentiviral based method for the rapid and sustained genetic manipulation of renal tubules. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Chen, P.; Zheng, P.; Yin, F.; Cheng, Q.; Zhou, Z.; Xie, H.; Li, J.; Ni, J.; Wang, Y.; et al. KLF4 initiates sustained YAP activation to promote renal fibrosis in mice after ischemia-reperfusion kidney injury. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Duan, X.; Zhu, W.; Liu, Y.; Wu, W.; Zeng, G. SaRNA-mediated activation of TRPV5 reduces renal calcium oxalate deposition in rat via decreasing urinary calcium excretion. Urolithiasis 2018, 46, 271–278. [Google Scholar] [CrossRef]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, 121ra18. [Google Scholar] [CrossRef] [Green Version]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Zhang, K.; Jiang, Z.Y.; Cai, L.H. MiR-204/miR-211 downregulation contributes to Candidemia-induced kidney injuries via derepression of Hmx1 expression. Life Sci. 2014, 102, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Invest. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Wang, J.; Miao, H. MiR-107 induces TNF-α secretion in endothelial cells causing tubular cell injury in patients with septic acute kidney injury. Biochem. Biophys. Res. Commun. 2017, 483, 45–51. [Google Scholar] [CrossRef]
- Wilflingseder, J.; Jelencsics, K.; Bergmeister, H.; Sunzenauer, J.; Regele, H.; Eskandary, F.; Reindl-Schwaighofer, R.; Kainz, A.; Oberbauer, R. miR-182-5p Inhibition Ameliorates Ischemic Acute Kidney Injury. Am. J. Pathol. 2017, 187, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Liu, Y.; Liu, P.; Hao, J.; Liang, M.; Mi, Q.S.; Chen, J.K.; Dong, Z. MicroRNA-489 induction by hypoxia-inducible factor-1 protects against ischemic kidney injury. J. Am. Soc. Nephrol. 2016, 27, 2784–2796. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Invest. 2018, 128, 5448–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.C.; Valencia, T.; Allerson, C.; Schairer, A.; Flaten, A.; Yheskel, M.; Kersjes, K.; Li, J.; Gatto, S.; Takhar, M.; et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat. Commun. 2019, 10, 4148. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Fu, J.; Wang, D.; Jiao, C.; Cui, X.; Chen, C.; Liu, D.; Zhang, Y.; Wang, Y.; Yuen, P.S.T.; et al. miR-150-Based RNA Interference Attenuates Tubulointerstitial Fibrosis through the SOCS1/JAK/STAT Pathway In vivo and In Vitro. Mol. Ther. Nucleic Acids 2020, 22, 871–884. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Lin, F.; Yin, H.; Li, X.; He, W.; Liu, N.; Gou, X. Exosomes from human-bone-marrow-derived mesenchymal stem cells protect against renal ischemia/reperfusion injury via transferring miR-199a-3p. J. Cell. Physiol. 2019, 234, 23736–23749. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Siemann, D.W. Inhibition of renal cell carcinoma angiogenesis and growth by antisense oligonucleotides targeting vascular endothelial growth factor. Br. J. Cancer 2002, 87, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Daniel, C.; Takabatake, Y.; Mizui, M.; Isaka, Y.; Kawashi, H.; Rupprecht, H.; Imai, E.; Hugo, C. Antisense oligonucleotides against thrombospondin-1 inhibit activation of TGF-β in fibrotic renal disease in the rat in vivo. Am. J. Pathol. 2003, 163, 1185–1192. [Google Scholar] [CrossRef]
- Kausch, I.; Jiang, H.; Brocks, C.; Bruderek, K.; Krüger, S.; Sczakiel, G.; Jocham, D.; Böhle, A. Ki-67-directed antisense therapy in an orthotopic renal cell carcinoma model. Eur. Urol. 2004, 46, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Xu, Z.; Tung, D.; Lanting, L.; Natarajan, R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007, 21, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Newbury, L.J.; Knisely, A.S.; Monia, B.; Hendry, B.M.; Sharpe, C.C. Antisense knockdown of Kras inhibits fibrosis in a rat model of unilateral ureteric obstruction. Am. J. Pathol. 2012, 180, 82–90. [Google Scholar] [CrossRef]
- Ravichandran, K.; Zafar, I.; He, Z.; Doctor, R.B.; Moldovan, R.; Mullick, A.E.; Edelstein, C.L. An mTOR anti-sense oligonucleotide decreases polycystic kidney disease in mice with a targeted mutation in Pkd2. Hum. Mol. Genet. 2014, 23, 4919–4931. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.; Ozkok, A.; Wang, Q.; Mullick, A.E.; Edelstein, C.L. Antisense-mediated angiotensinogen inhibition slows polycystic kidney disease in mice with a targeted mutation in Pkd2. Am. J. Physiol. Ren. Physiol. 2015, 308, F349–F357. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.M.; Park, K.K.; Lee, I.K.; Kim, J.K.; Kim, C.H.; Chang, Y.C. Ring-Sp1 decoy oligonucleotide effectively suppresses extracellular matrix gene expression and fibrosis of rat kidney induced by unilateral ureteral obstruction. Gene Ther. 2006, 13, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Tomita, N.; Kashihara, N.; Morishita, R. Transcription factor decoy oligonucleotide-based therapeutic strategy for renal disease. Clin. Exp. Nephrol. 2007, 11, 7–17. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.I. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Um, J.E.; Park, J.T.; Nam, B.Y.; Lee, J.P.; Jung, J.H.; Kim, Y.; Kim, S.; Park, J.; Wu, M.; Han, S.H.; et al. Periostin-binding DNA aptamer treatment attenuates renal fibrosis under diabetic conditions. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamagishi, S.I.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, s41598-s018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, Z.; Xie, L.; Zhang, Y.; Deng, T.; Li, J.; Liu, J.; Xiong, W.; Zhang, L.; Zhang, L.; et al. Molecular Recognition and In-Vitro-Targeted Inhibition of Renal Cell Carcinoma Using a DNA Aptamer. Mol. Ther. Nucleic Acids 2018, 12, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dammes, N.; Peer, D. Paving the Road for RNA Therapeutics. Trends Pharmacol. Sci. 2020, 41, 755–775. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, C.; Zhao, Z.; Zhu, T.; Yang, B. Fighting against kidney diseases with small interfering RNA: Opportunities and challenges. J. Transl. Med. 2015, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Glebova, K.; Reznik, O.N.; Reznik, A.O.; Mehta, R.; Galkin, A.; Baranova, A.; Skoblov, M. siRNA technology in kidney transplantation: Current status and future potential. BioDrugs 2014, 28, 345–361. [Google Scholar] [CrossRef]
- Kwok, A.; Raulf, N.; Habib, N. Developing small activating RNA as a therapeutic: Current challenges and promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef]
- Petrillo, F.; Iervolino, A.; Zacchia, M.; Simeoni, A.; Masella, C.; Capolongo, G.; Perna, A.; Capasso, G.; Trepiccione, F. MicroRNAs in Renal Diseases: A Potential Novel Therapeutic Target. Kidney Dis. 2017, 3, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Schena, F.P.; Serino, G.; Sallustio, F. MicroRNAs in kidney diseases: New promising biomarkers for diagnosis and monitoring. Nephrol. Dial. Transplant. 2014, 29, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledeganck, K.J.; Gielis, E.M.; Abramowicz, D.; Van Craenenbroeck, A.H.; Stenvinkel, P.; Shiels, P.G. MicroRNAs in AKI and kidney transplantation. Clin. J. Am. Soc. Nephrol. 2019, 14, 454–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburger, T.; Lorenzen, J.M. Diagnostic and Therapeutic Potential of microRNAs in Acute Kidney Injury. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, H.; Yheskel, M.; Patel, V. Modulation of polycystic kidney disease by non-coding RNAs. Cell. Signal. 2020, 71. [Google Scholar] [CrossRef]
- Li, D.; Sun, L. MicroRNAs and Polycystic Kidney Disease. Kidney Med. 2020, 2, 762–770. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, H.; Huang, Z.; Zheng, H.; Zhou, J. Emerging role of miRNAs in renal fibrosis. RNA Biol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Fan, F.; Wang, Y.; Gonzalez-Fernandez, E.; Wang, C.; Yang, L.; Booz, G.W.; Roman, R.J. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol. Genom. 2018, 50, 20–34. [Google Scholar] [CrossRef]
- Peters, L.J.F.; Floege, J.; Biessen, E.A.L.; Jankowski, J.; van der Vorst, E.P.C. Micrornas in chronic kidney disease: Four candidates for clinical application. Int. J. Mol. Sci. 2020, 21, 6547. [Google Scholar] [CrossRef] [PubMed]
- Sun, I.O.; Lerman, L.O. Urinary microRNA in kidney disease: Utility and roles. Am. J. Physiol. Ren. Physiol. 2019, 316, F785–F793. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Eswar, P.R.; Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, S.; Beal, J.; Ebrahimkhani, M.R.; Huh, J.; Hall, R.N.; Xie, Z.; Li, Y.; Weiss, R. CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Methods 2014, 11, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Cruz, N.M.; Freedman, B.S. CRISPR Gene Editing in the Kidney. Am. J. Kidney Dis. 2018, 71, 874–883. [Google Scholar] [CrossRef]
- Higashijima, Y.; Hirano, S.; Nangaku, M.; Nureki, O. Applications of the CRISPR-Cas9 system in kidney research. Kidney Int. 2017, 92, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Estrada, J.L.; Martens, G.; Li, P.; Adams, A.; Newell, K.A.; Ford, M.L.; Butler, J.R.; Sidner, R.; Tector, M.; Tector, J. Evaluation of human and non-human primate antibody binding to pig cells lacking GGTA1/CMAH/β4GalNT2 genes. Xenotransplantation 2015, 22, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Reyes, L.M.; Estrada, J.L.; Wang, Z.Y.; Blosser, R.J.; Smith, R.F.; Sidner, R.A.; Paris, L.L.; Blankenship, R.L.; Ray, C.N.; Miner, A.C.; et al. Creating Class I MHC–Null Pigs Using Guide RNA and the Cas9 Endonuclease. J. Immunol. 2014, 193, 5751–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higginbotham, L.; Mathews, D.; Breeden, C.A.; Song, M.; Farris, A.B.; Larsen, C.P.; Ford, M.L.; Lutz, A.J.; Tector, M.; Newell, K.A.; et al. Pre-transplant antibody screening and anti-CD154 costimulation blockade promote long-term xenograft survival in a pig-to-primate kidney transplant model. Xenotransplantation 2015, 22, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Cenni, E.; Perut, F.; Zuntini, M.; Granchi, D.; Amato, I.; Avnet, S.; Brandi, M.L.; Baldini, N. Inhibition of angiogenic activity of renal carcinoma by an antisense oligonucleotide targeting fibroblast growth factor-2-PubMed. Anticancer Res. 2005, 25, 1109–1113. [Google Scholar]
- Zellweger, T.; Miyake, H.; July, L.V.; Akbari, M.; Kiyama, S.; Gleave, M.E. Chemosensitization of human renal cell cancer using antisense oligonucleotides targeting the antiapoptotic gene clusterin. Neoplasia 2001, 3, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, M.; Menon, A.P.; Moreno, B.; Meraviglia-Crivelli, D.; Soldevilla, M.M.; Cartón-García, F.; Pastor, F. Aptamers Against Live Targets: Is In vivo SELEX Finally Coming to the Edge? Mol. Ther. Nucleic Acids 2020, 21, 192–204. [Google Scholar] [CrossRef]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Mondragón, E.; Maher, L.J. Anti-Transcription Factor RNA Aptamers as Potential Therapeutics. Nucleic Acid Ther. 2016, 26, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An rna toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Melone, M.A.B.; Montesarchio, D. Anti-VEGF DNA-based aptamers in cancer therapeutics and diagnostics. Med. Res. Rev. 2021, 41, 464–506. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Zhang, X. Aptamers as versatile ligands for biomedical and pharmaceutical applications. Int. J. Nanomedicine 2020, 15, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Nakamura, Y. Aptamers: A review of their chemical properties and modifications for therapeutic application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2020, 25, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranches, G.; Lukasser, M.; Schramek, H.; Ploner, A.; Stasyk, T.; Mayer, G.; Mayer, G.; Hüttenhofer, A. In Vitro Selection of Cell-Internalizing DNA Aptamers in a Model System of Inflammatory Kidney Disease. Mol. Ther. Nucleic Acids 2017, 8, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, Y.; Chen, Y.; Hong, S.; Sun, Y.; Sun, N.; Pei, R. In vitro selection of DNA aptamers against renal cell carcinoma using living cell-SELEX. Talanta 2017, 175, 235–242. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Invest. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G. Nucleic Acid Immunity. In Advances in Immunology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 133, pp. 121–169. [Google Scholar]
- Hecker, M.; Wagner, A.H. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017, 144, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Morales-Medina, J.; Palmieri, B. Phosphorothioate Oligonucleotides: Effectiveness and Toxicity. Curr. Drug Targets 2014, 15, 663–673. [Google Scholar] [CrossRef]
- Agrawal, S.; Teamani, J.; Galbraith, W.; Tang, J. Pharmacokinetics of Antisense Oligonucleotides. Clin. Pharmacokinet. 1995, 28, 7–16. [Google Scholar] [CrossRef]
- Kowalska, J.; Del Nogal, A.W.; Darzynkiewicz, Z.M.; Buck, J.; Nicola, C.; Kuhn, A.N.; Lukaszewicz, M.; Zuberek, J.; Strenkowska, M.; Ziemniak, M.; et al. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: Versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res. 2014, 42, 10245–10264. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Shen, W.; Crooke, S.T. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015, 43, 2927–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvam, C.; Mutisya, D.; Prakash, S.; Ranganna, K.; Thilagavathi, R. Therapeutic potential of chemically modified siRNA: Recent trends. Chem. Biol. Drug Des. 2017, 90, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; Liang, L.; McClintock, K.; Yaworski, E.; MacLachlan, I. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007, 15, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Bola, G.; Lee, A.C.H.; MacLachlan, I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006, 13, 494–505. [Google Scholar] [CrossRef]
- Zanardi, T.A.; Kim, T.W.; Shen, L.; Serota, D.; Papagiannis, C.; Park, S.Y.; Kim, Y.; Henry, S.P. Chronic Toxicity Assessment of 2′-O-Methoxyethyl Antisense Oligonucleotides in Mice. Nucleic Acid Ther. 2018, 28, 233–241. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009, 6, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, J.A. Comparative Renal Toxicopathology of Antisense Oligonucleotides. Nucleic Acid Ther. 2016, 26, 199–209. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Janssen, M.J.; Nieskens, T.T.G.; Steevels, T.A.M.; Caetano-Pinto, P.; den Braanker, D.; Mulder, M.; Ponstein, Y.; Jones, S.; Masereeuw, R.; den Besten, C.; et al. Therapy with 2′-O-Me Phosphorothioate Antisense Oligonucleotides Causes Reversible Proteinuria by Inhibiting Renal Protein Reabsorption. Mol. Ther. Nucleic Acids 2019, 18, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Frazier, K.S.; Obert, L.A. Drug-induced Glomerulonephritis: The Spectre of Biotherapeutic and Antisense Oligonucleotide Immune Activation in the Kidney. Toxicol. Pathol. 2018, 46, 904–917. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Rubin, J.D.; Barry, M.A. Improving Molecular Therapy in the Kidney. Mol. Diagnosis Ther. 2020, 24, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.J.; Cherqui, S. Gene transfer to mouse kidney in vivo. Methods Mol. Biol. 2019, 1937, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.D.; Nguyen, T.V.; Allen, K.L.; Ayasoufi, K.; Barry, M.A. Comparison of Gene Delivery to the Kidney by Adenovirus, Adeno-Associated Virus, and Lentiviral Vectors after Intravenous and Direct Kidney Injections. Hum. Gene Ther. 2019, 30, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Park, F. Gene therapy research for kidney diseases. Physiol. Genom. 2019, 51, 449–461. [Google Scholar] [CrossRef]
- Rocca, C.J.; Ur, S.N.; Harrison, F.; Cherqui, S. RAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: Conclusion of a comparative study. Gene Ther. 2014, 21, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.C.; Fogelgren, B.; Park, K.M.; Heidenberg, J.; Zuo, X.; Huang, L.; Bennett, J.; Lipschutz, J.H. Adeno-Associated Virus-Mediated Gene Transfer to Renal Tubule Cells via a Retrograde Ureteral Approach. Nephron Extra 2011, 1, 217–223. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Huang, Z. Recent progress in microRNA-based delivery systems for the treatment of human disease. ExRNA 2019, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug. Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Asico, L.D.; Cuevas, S.; Ma, X.; Jose, P.A.; Armando, I.; Konkalmatt, P.R. Nephron segment-specific gene expression using AAV vectors. Biochem. Biophys. Res. Commun. 2018, 497, 19–24. [Google Scholar] [CrossRef]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Hanai, K.; Kojima, T.; Ota, M.; Onodera, J.; Sawada, N. Effects of Atelocollagen Formulation Containing Oligonucleotide on Endothelial Permeability. J. Drug Deliv. 2012, 2012, 245835. [Google Scholar] [CrossRef] [Green Version]
- Wada, F.; Yamamoto, T.; Ueda, T.; Sawamura, M.; Wada, S.; Harada-Shiba, M.; Obika, S. Cholesterol-GalNAc dual conjugation strategy for reducing renal distribution of antisense oligonucleotides. Nucleic Acid Ther. 2018, 28, 50–57. [Google Scholar] [CrossRef]
- Shajari, N.; Mansoori, B.; Davudian, S.; Mohammadi, A.; Baradaran, B. Overcoming the Challenges of siRNA Delivery: Nanoparticle Strategies. Curr. Drug Deliv. 2016, 14, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; Van Der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Kang, P.M. Recent advances in nanocarrier-assisted therapeutics delivery systems. Pharmaceutics 2020, 12, 837. [Google Scholar] [CrossRef]
- Ma, Y.; Cai, F.; Li, Y.; Chen, J.; Han, F.; Lin, W. A review of the application of nanoparticles in the diagnosis and treatment of chronic kidney disease. Bioact. Mater. 2020, 5, 732–743. [Google Scholar] [CrossRef]
- Han, S.J.; Williams, R.M.; D’Agati, V.; Jaimes, E.A.; Heller, D.A.; Lee, H.T. Selective nanoparticle-mediated targeting of renal tubular Toll-like receptor 9 attenuates ischemic acute kidney injury. Kidney Int. 2020, 98, 76–87. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Riccardi, C.; Fàbrega, C.; Grijalvo, S.; Vitiello, G.; D’Errico, G.; Eritja, R.; Montesarchio, D. AS1411-decorated niosomes as effective nanocarriers for Ru(III)-based drugs in anticancer strategies. J. Mater. Chem. B 2018, 6, 5368–5384. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, A.; You, J.; Li, J.; Xu, H.; Xu, K. Fabrication of AS1411 aptamer functionalized Gd2O3-based molecular magnetic resonance imaging (mMRI) nanoprobe for renal carcinoma cell imaging. RSC Adv. 2015, 5, 77204–77210. [Google Scholar] [CrossRef]
- Li, J.; You, J.; Dai, Y.; Xu, K. Preparation of GO/BSA-Gd2O3/AS1411-DOX theranostic nanocomplex and its application in MRI study of Renal cell carcinoma. J. Nucl. Med. 2015, 56, 1035. [Google Scholar]
- Kelemen, R.E.; Mukherjee, R.; Cao, X.; Erickson, S.B.; Zheng, Y.; Chatterjee, A. A Precise Chemical Strategy To Alter the Receptor Specificity of the Adeno-Associated Virus. Angew. Chem. Int. Ed. 2016, 55, 10645–10649. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, R.E.; Erickson, S.B.; Chatterjee, A. Production and Chemoselective Modification of Adeno- Associated Virus Site-Specifically Incorporating an Unnatural Amino Acid Residue into Its Capsid. Noncanonical. Amin. Acids Methods Protoc. Methods Mol. Biol. 2018, 1728, 313–326. [Google Scholar]
- Katrekar, D.; Moreno, A.M.; Chen, G.; Worlikar, A.; Mali, P. Oligonucleotide conjugated multi-functional adeno-associated viruses. Sci. Rep. 2018, 8, 3589. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, X.; Di, B.Y.; Wang, D.; Zhang, Y.; Xia, H.; Mao, Q. Aptamer modification improves the adenoviral transduction of malignant glioma cells. J. Biotechnol. 2013, 168, 362–366. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, L.; Cui, C.; Cansiz, S.; Liang, H.; Wu, C.; Teng, I.T.; Chen, W.; Liu, Y.; Hou, W.; et al. Enhanced Targeted Gene Transduction: AAV2 Vectors Conjugated to Multiple Aptamers via Reducible Disulfide Linkages. J. Am. Chem. Soc. 2018, 140, 2–5. [Google Scholar] [CrossRef] [PubMed]
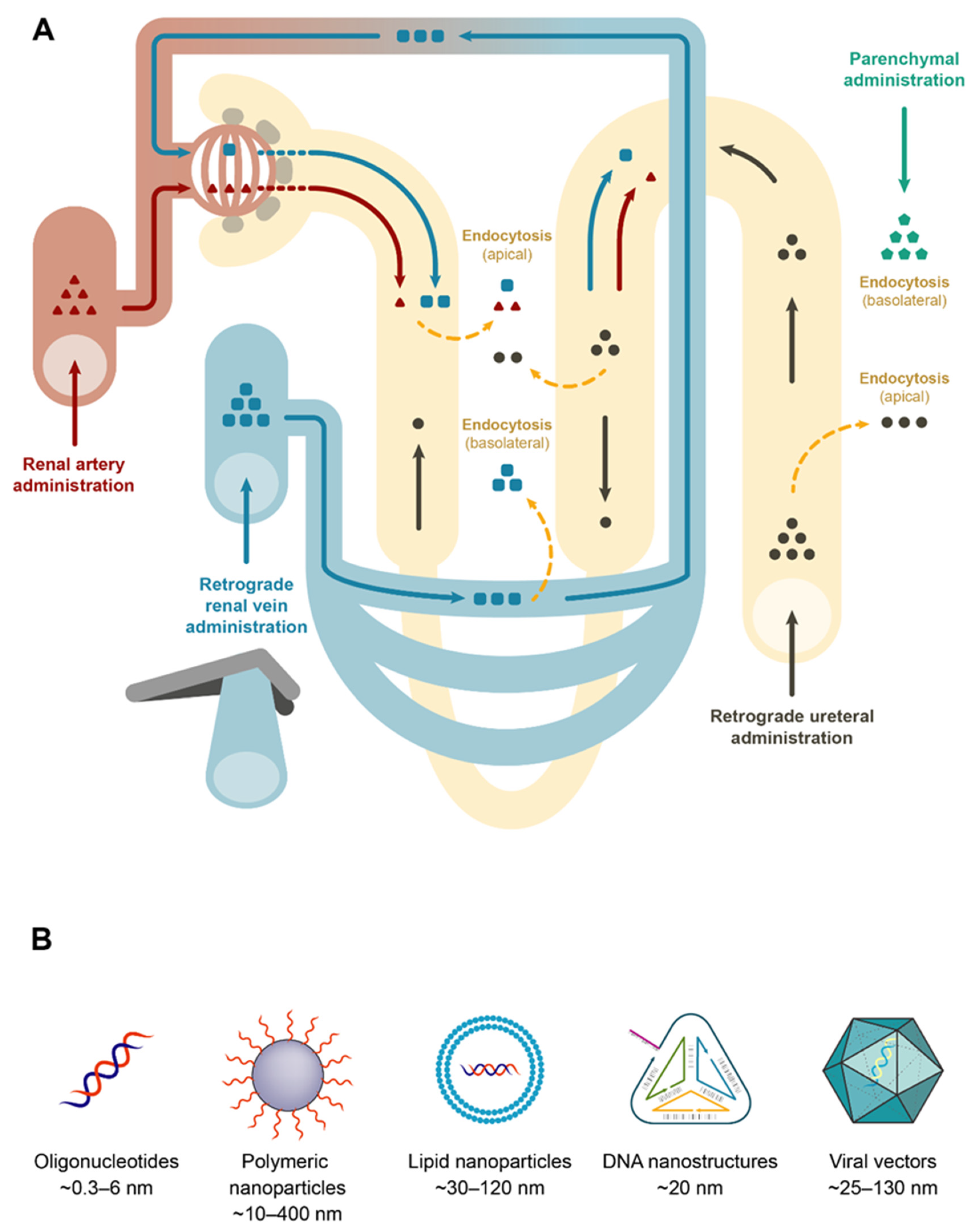

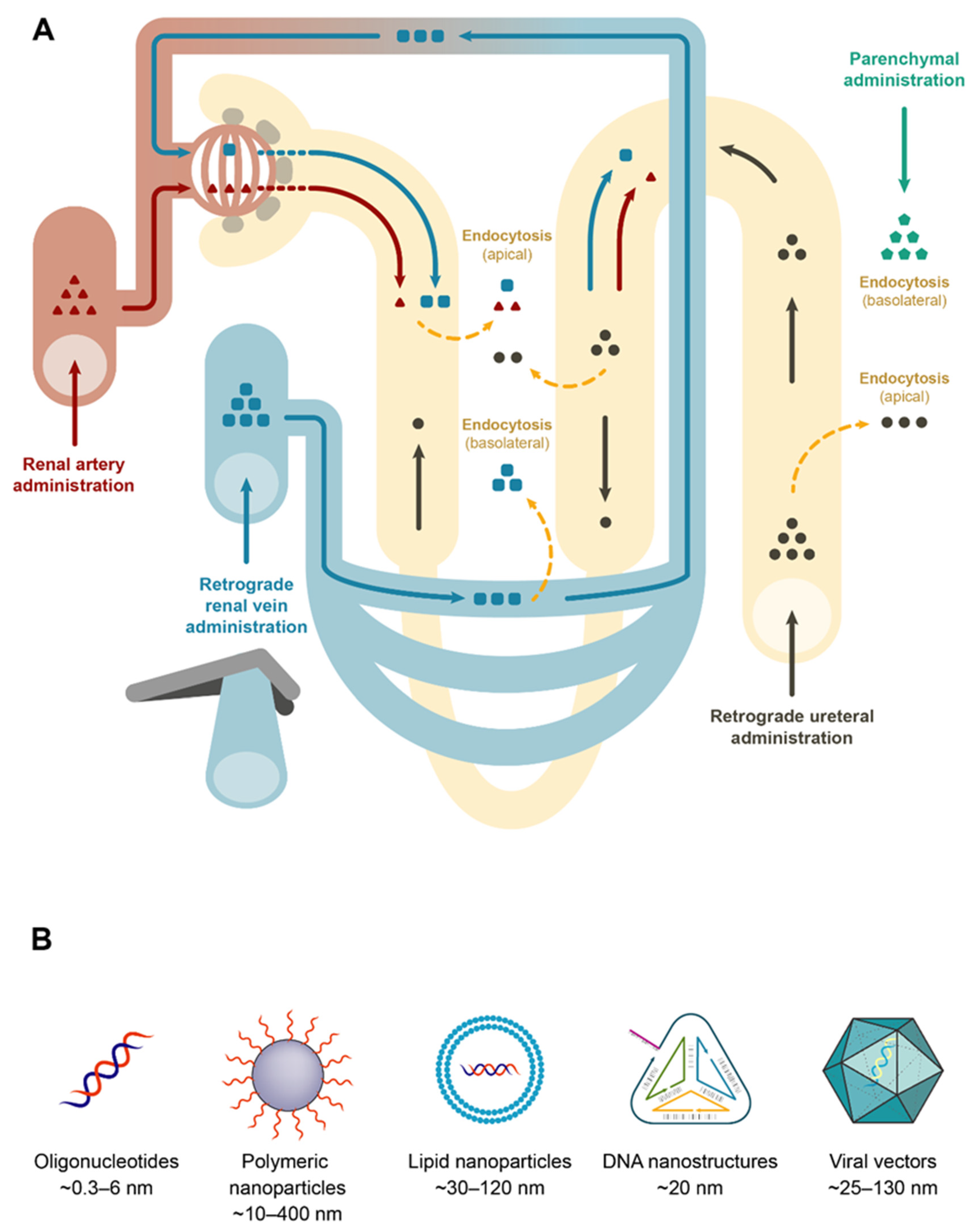{kind=link}
| STRATEGY | REFERENCE | RENAL TARGETS (Target; Cell/Tissue Type; Animal Model) | SEQUENCE (5′-3′) | CARRIER AND ROUTE OF ADMINISTRATION |
|---|---|---|---|---|
| siRNA | Molitoris et al., 2009 [15] | Trp53; PTECs; cisplatin-induced and ischemic AKI models in rats | GAAGAAAAUUUCCGCAAAA | Naked; IV |
| Takabatake et al., 2009 [16] | Egfp and Tgfb1; glomeruli; glomerulonephritis model in rats | Egfp—GGCUACGUCCAGGAGCGCA Tgfb1—GUCAACUGUGGAGCAACACdTdT | Naked, RA | |
| Shimizu et al., 2010 [17] | Mapk1; glomeruli; glomerulonephritis model in mice and rats | UGCUGACUCCAAAGCUCUGdTdT | Polyion complex nanocarriers; IP | |
| Gao et al., 2014 [18] | Aqp1; PTECs; mice | CGCAACUUCUCAAACCACUTT | Chitosan NPs; IV | |
| Morishita et al., 2014 [19] | Smad4; tubulointerstitium and tubule epithelial cells; renal fibrosis model in mice | GAUGAAUUGGAUUCUUUAATT | Naked; IV | |
| Yang et al., 2015 [20] | Cox2; peritoneal macrophages recruited to the kidney; UUO model in mice | GGAUUUGACCAGUAUAAGUTT | Chitosan NPs; IP | |
| Zuckerman et al., 2015 [21] | Egfp; glomerular mesangium; mice | GGCUACGUCCAGGAGCGCACC | Polycationic cyclodextrin NPs functionalized with mannose and transferrin; IV | |
| Zheng et al., 2016 [22] | Fas, C3 and RelB; glomeruli and medullar tubule cells; ischemic AKI in mice | Fas—GUGCAAGUGCAA ACCAGAC C3—GUGCAAGACUUCCUAAAGA RelB—GGAAUCGAGAGCAAACGAA | Naked; RA | |
| Alidori et al., 2016 [23] | Ctr1, Trp53 and Mep1b; cortex and PTECs; AKI model in mice | Ctr1—GGCAUGAACAUGUGAAUUGCUGGTT Trp53—AGGAGUCACAGUCGGAUAUCAGCCT Mep1b—GGAAUUGACCAAGACAUAUUU GATA | Fibrillar carbon nanotubes (fCNT); IV | |
| Eadon et al., 2017 [24] | Lrp2; PTECs, mice | Naked or lipid-base transfection; IV | ||
| Narváez et al., 2019 [25] | Cd40; tubulointerstitium; UUO model in mice | GUGUGUUACGUGCAGUGACUU | Naked; IV | |
| Wang et al., 2020 [26] | p38α MapK and p65; glomerular mesangium and peritubular endothelial cells; glomerulonephritis model in mice | p38α—GGUCACUGGAGGAAUUC p65—GCGACAAGGUGCAGAAAGA | Liposomal NPs, IV | |
| Thai et al., 2020 [27] | Trp53; tubular epithelial cells; AKI model in mice | GAGAAUAUUUCACCCUUCA | DNA nanostructure; IV | |
| shRNA plasmid | Wang et al., 2006 [28] | Mr; cortical tubule cells; renal hypertension and damage model in rats | CCAACAAGGAAGCCTGAGC | AAV; IV |
| Shou et al., 2009 [29] | Sharp2; T-cells; transplantation model in rats | ACCCGAACATCTCAAACTTA | Lentivirus; ex vivo perfusion | |
| Zhou et al., 2011 [30] | ColI; cortex; rats | GCAACCTGGATGCCATCAA | Lentivirus; RP | |
| Fujino et al., 2013 [31] | Trp53: cortex and medullar tubule cells; ischemic AKI model in mice | Cationic polymer; RA | ||
| Espana-Agusti et al., 2015 [32] | Tsc1 and Luc; PTECs, DTECs and interstitium; mice | Tsc1—CGGAAGAAGCTGCAATATCTAA Luc—CCGCCTGAAGTCTCTGATTAA | Lentivirus; RP | |
| Xu et al., 2020 [33] | Yap and Klf4; renal tubules; ischemic AKI model in mice | Adenovirus; RP | ||
| saRNA | Zeng et al., 2018 [34] | Trpv5; medullar tubule cells; calcium crystal formation model in rats | AAGGGTCTCATGATTTCTCTA | Naked; RU |
| miRNA antagomir | Chau et al., 2012 [35] | miR21; PTECs; UUO model in mice | Naked; IP | |
| Putta et al., 2012 [36] | miR192; cortex and glomeruli; DN model in mice | GGCTGTCAATTCATAGGTCAG | Naked; SC | |
| Li et al., 2014 [37] | miR204; cortex and medulla; candidemia-induced AKI model in mice | AGGCAUAGGAUGACAAAGGGAA | Naked; IV | |
| Gomez et al., 2015 [38] | miR21; PTECs, Alport nephropathy mouse model | Naked, SC | ||
| Wang et al., 2017 [39] | miR107; peritubular endothelial cells; septic AKI model in mice | Complexed with atelocollagen; IV | ||
| Wilflingseder et al., 2017 [40] | miR182-5p; cortex and medulla; ischemic AKI model in mice, rats and pigs | Naked; IV (mice and rats), ex vivo perfusion (pig) | ||
| Wei et al., 2016 [41] | miR489; tubular epithelial cells; ischemic AKI model in mice | Naked; IV | ||
| Wei et al., 2018 [42] | miR668; tubular epithelial cells; ischemic AKI model in mice | Naked; IV | ||
| Lee et al., 2019 [43] | miR17; PTECs; ADPKD mouse model | GUUUCACGA | Naked; SC | |
| Luan et al., 2020 [44] | miR150; cortex and medulla; renal fibrosis model in mice | UACAAGGGUUGGGAG | Naked; IV | |
| miRNA mimic | Li et al., 2014 [37] | miR204 and miR211; cortex and medulla; candidemia-induced AKI in mice | miR204—UCCCGGUAAUCCCUUACCUGGUU CCCUUCCUU miR211—UCCCGGCUUUCCCUUACCUGGUUUUCCCCCUU | Naked, IV |
| Wei et al., 2018 [42] | miR668; cortex and medulla; ischemic AKI model in mice | Lipid-based transfection reagent; IV | ||
| Zhu et al., 2019 [45] | miR199a-3p; tubular epithelial cells; ischemic model AKI in mice | Exosomes; IV | ||
| ASO | Shi and Siemann [46] | Vegf; Caki-I RCC cell line; xenograft model in mice | CTCACCCGTCCATGAGCCCG | Naked; IV |
| Daniel et al., 2003 [47] | Tsp1; glomeruli; glomerulonephritis model in mice | Tsp1-1—TTCTCCGTTGTGATTGAA Tsp1-2—CACCTCCAATGAGTT | Naked by electroporation or HVJ-liposomes; RA | |
| Kausch et al., 2004 [48] | Ki67; Renca cells; RCC orthotopic model in mice | ACCAGGTGAGCCGAGGACGCCAT | Naked, IP | |
| Guha et al., 2007 [49] | Ctgf; PTECs and mesangial cells; DN model in mice | CCACAAGCTGTCCAGTCTAA | Naked; SC | |
| Wang et al., 2012 [50] | Kras; tubular epithelial cells; UUO model in rats | Kras-1—ATTCACATGACTATACACCT Kras-2—CACACTTATTCCCTACTAGG | Naked; SC | |
| Ravichandran et al., 2014 [51] | mTORC; tubular epithelial cells; PKD mouse model | TCCACTTTTCACAGCACTGC | Naked, IP | |
| Ravichandran et al., 2015 [52] | Agt; tubular epithelial cells; PKD mouse model | TCTTCCACCCTGTCACAGCC | Naked, IP | |
| TFD | Chae et al., 2006 [53] | Sp1; tubulointerstitium; UUO model in rats | GGGGCGGGGC | HVJ-liposomes; RV |
| Tomita et al., 2007 [54] | E2f; glomeruli; rats | HVJ-liposomes; RA | ||
| Aptamers | Matsui et al., 2017 [55] | RAGE; kidney, heart, eyes, testis; DN model in rats | CCTGATATGGTGTCACCGCCGCCTTAGTATTGGTGTCTAC | Naked; IP |
| Um et al., 2017 [56] | Periostin; medulla; DN model in mice | PEG-conjugated; IP | ||
| Taguchi et al., 2018 [57] | RAGE; glomeruli; hypertensive mouse model | CATTCTTAGATTTTTGTCTCACTTAGGTGTAGATGGTGAT | Naked; SC | |
| Zhang et al., 2018 [58] | RCC 786-O cells; xenograft model in mice | ACTCATAGGGTTAGGGGCTGCTGGCCAGATATTCAGATGGTAGGGTTACTATGA | Naked; IV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cartón-García, F.; Saande, C.J.; Meraviglia-Crivelli, D.; Aldabe, R.; Pastor, F. Oligonucleotide-Based Therapies for Renal Diseases. Biomedicines 2021, 9, 303. https://doi.org/10.3390/biomedicines9030303
Cartón-García F, Saande CJ, Meraviglia-Crivelli D, Aldabe R, Pastor F. Oligonucleotide-Based Therapies for Renal Diseases. Biomedicines. 2021; 9(3):303. https://doi.org/10.3390/biomedicines9030303
Chicago/Turabian StyleCartón-García, Fernando, Cassondra Jeanette Saande, Daniel Meraviglia-Crivelli, Rafael Aldabe, and Fernando Pastor. 2021. "Oligonucleotide-Based Therapies for Renal Diseases" Biomedicines 9, no. 3: 303. https://doi.org/10.3390/biomedicines9030303
APA StyleCartón-García, F., Saande, C. J., Meraviglia-Crivelli, D., Aldabe, R., & Pastor, F. (2021). Oligonucleotide-Based Therapies for Renal Diseases. Biomedicines, 9(3), 303. https://doi.org/10.3390/biomedicines9030303