
Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets?


Abstract
:1. Introduction
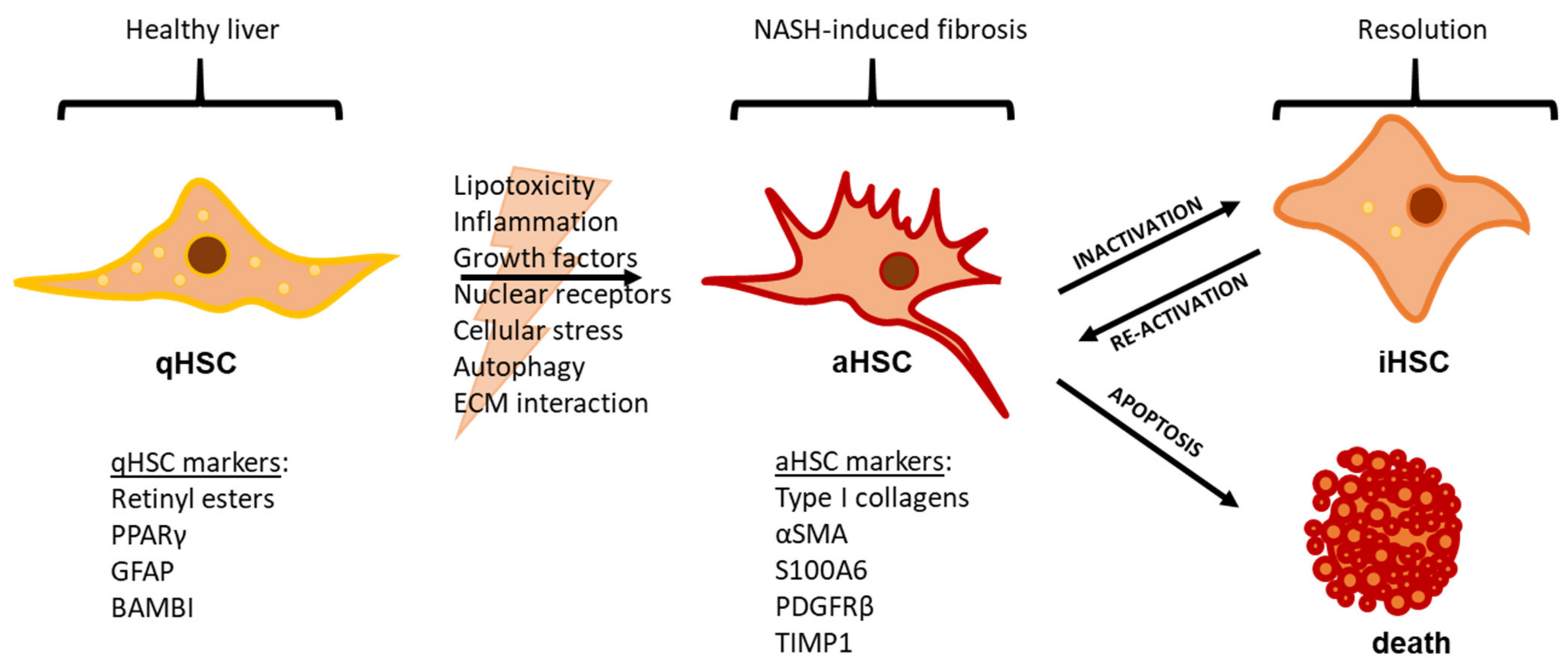
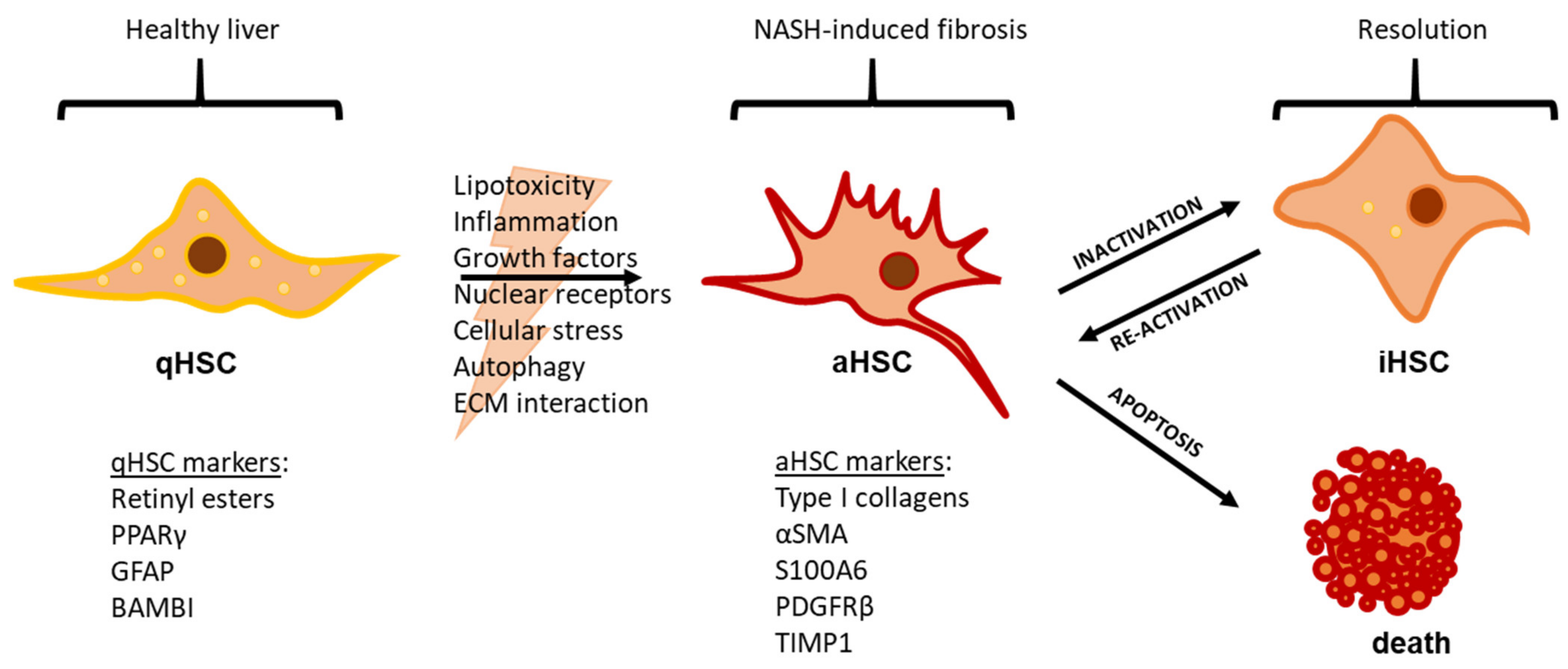
2. NAFLD Etiology and the Role of Hepatic Stellate Cells
3. Mechanisms of HSC Activation
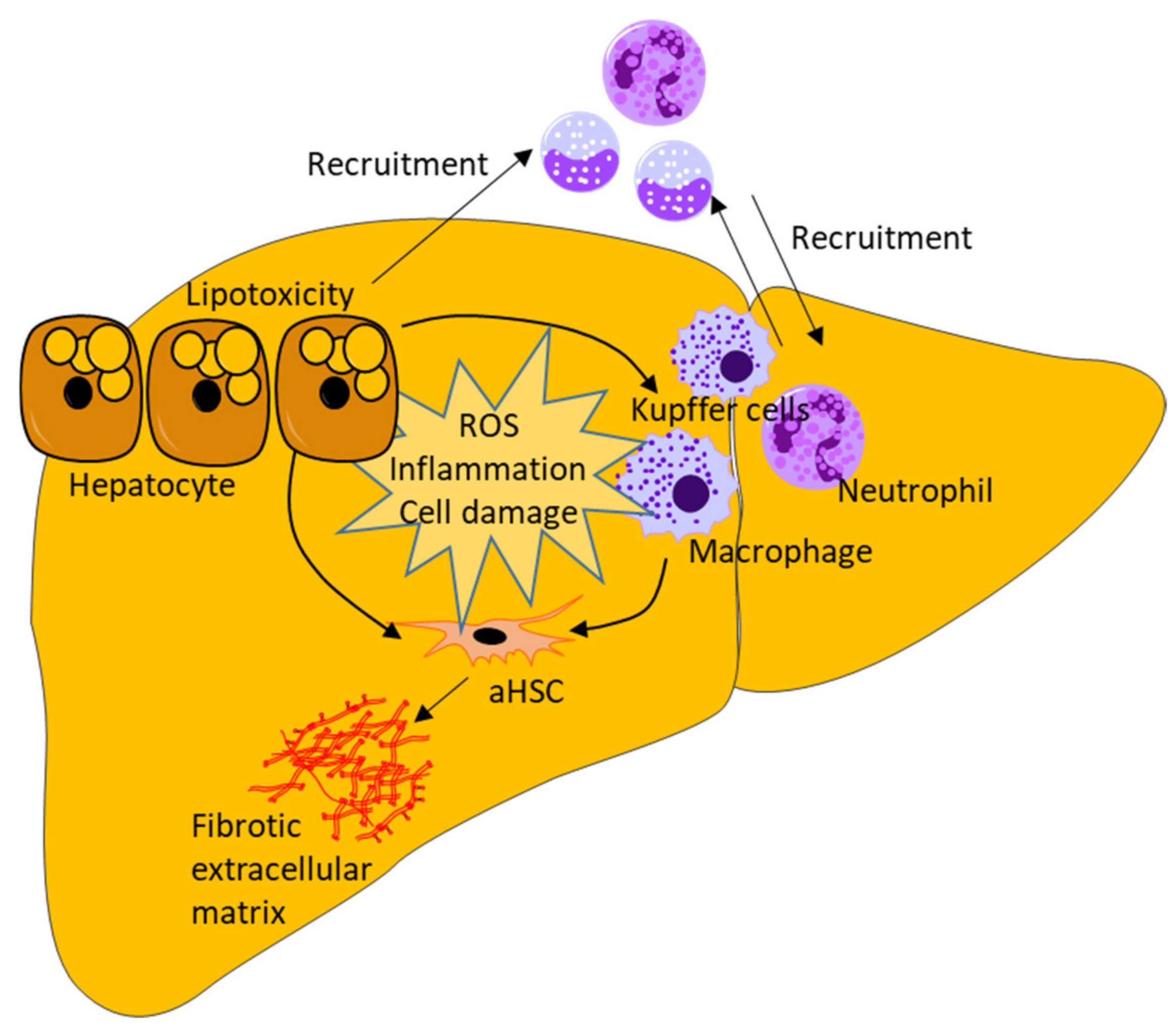
3.1. Lipotoxicity and Inflammation
3.2. Growth Factors
3.3. Nuclear Receptors
3.4. Cellular Stress and Autophagy
4. HSC Inactivation and Apoptosis
5. Pharmacotherapies with Putative Effects on HSCs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wai-Sun Wong, V.; Peleg, N.; et al. Association Between Fibrosis Stage and Outcomes of Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625.e1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Karin, D.; Koyama, Y.; Brenner, D.; Kisseleva, T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation 2016, 92, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoinne, S.; Friedman, S.L. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr. Opin. Pharmacol. 2019, 49, 60–70. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The Diagnosis and Management of Non-alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedossa, P. Histological Assessment of NAFLD. Dig. Dis. Sci. 2016, 61, 1348–1355. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Normal weight dyslipidemia: Is it all about the liver? Obes. Silver Spring 2016, 24, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, R.G. The portal fibroblast: Not just a poor man’s stellate cell. Gastroenterology 2014, 147, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef]
- Wake, K. ”Sternzellen” in the liver: Perisinusoidal cells with special reference to storage of vitamin A. Am. J. Anat. 1971, 132, 429–462. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Mallat, A.; Lotersztajn, S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am. J. Physiol. Cell Physiol. 2013, 305, C789–C799. [Google Scholar] [CrossRef] [Green Version]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef]
- Wilson, C.L.; Mann, J.; Walsh, M.; Perrugoria, M.J.; Oakley, F.; Wright, M.C.; Brignole, C.; Di Paolo, D.; Perri, P.; Ponzoni, M.; et al. Quiescent hepatic stellate cells functionally contribute to the hepatic innate immune response via TLR3. PLoS ONE 2014, 9, e83391. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- El Taghdouini, A.; Sørensen, A.L.; Reiner, A.H.; Coll, M.; Verhulst, S.; Mannaerts, I.; Øie, C.I.; Smedsrød, B.; Najimi, M.; Sokal, E.; et al. Genome-wide analysis of DNA methylation and gene expression patterns in purified, uncultured human liver cells and activated hepatic stellate cells. Oncotarget 2015, 6, 26729–26745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728.e1714–1744.e1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, A.L.; Schilling, J.D.; King, K.R.; Feldstein, A.E. The Power of Single Cell Analysis for the Study of Liver Pathobiology. Hepatology 2020, 73, 437–448. [Google Scholar] [CrossRef]
- Deleve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Martinez-Hernandez, A.; Martinez, J. The role of capillarization in hepatic failure: Studies in carbon tetrachloride-induced cirrhosis. Hepatology 1991, 14, 864–874. [Google Scholar] [CrossRef]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef]
- Iredale, J.P.; Benyon, R.C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J.P. Mechanisms of spontaneous resolution of rat liver fibrosis-Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 1998, 102, 538–549. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847.e1838. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Wu, N.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Venter, J.; Standeford, H.; Onori, P.; Marzioni, M.; Alvaro, D.; Franchitto, A.; et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-β1 biliary secretion in mice. Hepatology 2016, 64, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Iredale, J.P. Hepatic stellate cell behavior during resolution of liver injury. Semin. Liver Dis 2001, 21, 427–436. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.Y.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083 e1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164 e110. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamura, T.; Fujisawa, T.; Husain, S.R.; Kioi, M.; Nakajima, A.; Puri, R.K. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 2008, 181, 4656–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Meyer, C.; Muller, A.; Herweck, F.; Li, Q.; Mullenbach, R.; Mertens, P.R.; Dooley, S.; Weng, H.L. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J. Immunol. 2011, 187, 2814–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichs, D.; Berres, M.L.; Nellen, A.; Fischer, P.; Scholten, D.; Trautwein, C.; Wasmuth, H.E.; Sahin, H. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS ONE 2013, 8, e66106. [Google Scholar] [CrossRef] [PubMed]
- Berres, M.L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Invest 2010, 120, 4129–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: A cross-sectional study. Sci. Rep. 2014, 4, 5832. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Conca, P.; Riccio, A.; Tarantino, M.; Di Minno, M.N.; Chianese, D.; Pasanisi, F.; Contaldo, F.; Scopacasa, F.; Capone, D. Enhanced serum concentrations of transforming growth factor-beta1 in simple fatty liver: Is it really benign? J. Transl. Med. 2008, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda-Flores, R.N.; Vera-Cabrera, L.; Flores-Gutierrez, J.P.; Maldonado-Garza, H.; Salinas-Garza, R.; Zorrilla-Blanco, P.; Bosques-Padilla, F.J. Obesity-related non-alcoholic steatohepatitis and TGF-beta1 serum levels in relation to morbid obesity. Ann. Hepatol. 2002, 1, 36–39. [Google Scholar] [CrossRef]
- Nair, B.; Nath, L.R. Inevitable role of TGF-β1 in progression of nonalcoholic fatty liver disease. J. Recept Signal Transduct. Res. 2020, 40, 195–200. [Google Scholar] [CrossRef]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Nieto, N.; Dominguez-Rosales, J.A.; Fontana, L.; Salazar, A.; Armendariz-Borunda, J.; Greenwel, P.; Rojkind, M. Rat hepatic stellate cells contribute to the acute-phase response with increased expression of alpha1(I) and alpha1(IV) collagens, tissue inhibitor of metalloproteinase-1, and matrix-metalloproteinase-2 messenger RNAs. Hepatology 2001, 33, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Truter, S.; Greenwel, P.; Rojkind, M.; Unoura, M.; Kobayashi, K.; Ramirez, F. Regulation of the alpha 2(I) collagen gene transcription in fat-storing cells derived from a cirrhotic liver. Hepatology 1995, 22, 573–579. [Google Scholar]
- Herbst, H.; Wege, T.; Milani, S.; Pellegrini, G.; Orzechowski, H.D.; Bechstein, W.O.; Neuhaus, P.; Gressner, A.M.; Schuppan, D. Tissue inhibitor of metalloproteinase-1 and -2 RNA expression in rat and human liver fibrosis. Am. J. Pathol. 1997, 150, 1647–1659. [Google Scholar] [PubMed]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yafnagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yamagata, H.; Furukawa, F.; Seki, T.; Nishizawa, M.; Fujisawa, J.; Okazaki, K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 2005, 166, 1029–1039. [Google Scholar] [CrossRef]
- Dooley, S.; Delvoux, B.; Lahme, B.; Mangasser-Stephan, K.; Gressner, A.M. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 2000, 31, 1094–1106. [Google Scholar] [CrossRef]
- Tahashi, Y.; Matsuzaki, K.; Date, M.; Yoshida, K.; Furukawa, F.; Sugano, Y.; Matsushita, M.; Himeno, Y.; Inagaki, Y.; Inoue, K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 2002, 35, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kang, J.H.; Choi, S.Y.; Suk, K.T.; Kim, D.J.; Kwon, O.S. PKCdelta as a regulator for TGFbeta1-induced alpha-SMA production in a murine nonalcoholic steatohepatitis model. PLoS ONE 2013, 8, e55979. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef]
- Walter, R.; Wanninger, J.; Bauer, S.; Eisinger, K.; Neumeier, M.; Weiss, T.S.; Amann, T.; Hellerbrand, C.; Schaffler, A.; Scholmerich, J.; et al. Adiponectin reduces connective tissue growth factor in human hepatocytes which is already induced in non-fibrotic non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2011, 91, 740–744. [Google Scholar] [CrossRef]
- Colak, Y.; Senates, E.; Coskunpinar, E.; Oltulu, Y.M.; Zemheri, E.; Ozturk, O.; Doganay, L.; Mesci, B.; Yilmaz, Y.; Enc, F.Y.; et al. Concentrations of connective tissue growth factor in patients with nonalcoholic fatty liver disease: Association with liver fibrosis. Dis. Markers 2012, 33, 77–83. [Google Scholar] [CrossRef]
- Sakai, K.; Jawaid, S.; Sasaki, T.; Bou-Gharios, G.; Sakai, T. Transforming growth factor-beta-independent role of connective tissue growth factor in the development of liver fibrosis. Am. J. Pathol. 2014, 184, 2611–2617. [Google Scholar] [CrossRef]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front Biosci. (Landmark Ed.) 2012, 17, 2495–2507. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Xie, Y.; Peng, M.; Ma, L.; Zhou, Y.; Zhang, Y.; Kang, W.; Wang, J.; Bai, X.; Wang, P.; et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin. Exp. Med. 2014, 14, 141–150. [Google Scholar] [CrossRef]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Czochra, P.; Klopcic, B.; Meyer, E.; Herkel, J.; Garcia-Lazaro, J.F.; Thieringer, F.; Schirmacher, P.; Biesterfeld, S.; Galle, P.R.; Lohse, A.W.; et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J. Hepatol. 2006, 45, 419–428. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef]
- Lambrecht, J.; Verhulst, S.; Mannaerts, I.; Sowa, J.-P.; Best, J.; Canbay, A.; Reynaert, H.; van Grunsven, L.A. A PDGFRβ-based score predicts significant liver fibrosis in patients with chronic alcohol abuse, NAFLD and viral liver disease. EBioMedicine 2019, 43, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Invest. 1994, 94, 1563–1569. [Google Scholar] [CrossRef] [Green Version]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.-C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Brown, K.D.; Witek, R.P.; Omenetti, A.; Yang, L.; Vandongen, M.; Milton, R.J.; Hines, I.N.; Rippe, R.A.; Spahr, L.; et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 2008, 134, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.V.; Michelotti, G.A.; Pereira Tde, A.; Boursier, J.; Kruger, L.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2015, 64, 1148–1157. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Li, Y.X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab. Invest. 2005, 85, 1368–1380. [Google Scholar] [CrossRef]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab. Invest. 2007, 87, 499–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, W.K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488 e1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Crn, N. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Kruger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Invest. 2013, 123, 2380–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsova, P.; Ibrahim, S.H.; Bronk, S.F.; Yagita, H.; Gores, G.J. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e70599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Grunsven, L.A. 3D in vitro models of liver fibrosis. Adv. Drug. Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; He, Y.P.; Zhao, H.; Xu, X.W. Hypoxia inducible factor-1 promotes liver fibrosis in nonalcoholic fatty liver disease by activating PTEN/p65 signaling pathway. J. Cell Biochem. 2019, 120, 14735–14744. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.D.; Traussnigg, S.A.; Trauner, M. Nuclear Receptor Modulation for the Treatment of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2016, 36, 69–86. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Xiong, S.; Hazra, S.; Tsukamoto, H. Adipogenic transcriptional regulation of hepatic stellate cells. J. Biol. Chem. 2005, 280, 4959–4967. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Deng, X.; Zhai, X.; Zhou, M.; Jia, X.; Luo, L.; Niu, M.; Zhu, H.; Qiang, H.; Zhou, Y. p38 mitogen-activated protein kinase and liver X receptor-alpha mediate the leptin effect on sterol regulatory element binding protein-1c expression in hepatic stellate cells. Mol. Med. 2012, 18, 10–18. [Google Scholar] [CrossRef]
- Beaven, S.W.; Wroblewski, K.; Wang, J.H.; Hong, C.; Bensinger, S.; Tsukamoto, H.; Tontonoz, P. Liver X Receptor Signaling Is a Determinant of Stellate Cell Activation and Susceptibility to Fibrotic Liver Disease. Gastroenterology 2011, 140, 1052–1062. [Google Scholar] [CrossRef] [Green Version]
- Moran-Salvador, E.; Titos, E.; Rius, B.; Gonzalez-Periz, A.; Garcia-Alonso, V.; Lopez-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Claria, J. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef]
- Nan, Y.M.; Han, F.; Kong, L.B.; Zhao, S.X.; Wang, R.Q.; Wu, W.J.; Yu, J. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand. J. Gastroenterol. 2011, 46, 358–369. [Google Scholar] [CrossRef]
- Yao, J.; Zhou, C.S.; Ma, X.; Fu, B.Q.; Tao, L.S.; Chen, M.; Xu, Y.P. FXR agonist GW4064 alleviates endotoxin-induced hepatic inflammation by repressing macrophage activation. World J. Gastroenterol. 2014, 20, 14430–14441. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Morelli, A.; Pruzanski, M.; Pellicciari, R. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor γ contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J. Pharmacol. Exp. Ther. 2005, 315, 58–68. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pruzanski, M.; Morelli, A.; Pellicciari, R. A farnesoid x receptor-small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J. Pharmacol. Exp. Ther. 2005, 314, 584–595. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, J.; Liu, Q.; Harnish, D.C. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 380–388. [Google Scholar] [CrossRef]
- Kong, B.; Luyendyk, J.P.; Tawfik, O.; Guo, G.L. Farnesoid X Receptor Deficiency Induces Nonalcoholic Steatohepatitis in Low-Density Lipoprotein Receptor-Knockout Mice Fed a High-Fat Diet. J. Pharmacol. Exp. Ther. 2009, 328, 116–122. [Google Scholar] [CrossRef]
- Jung, D.; Mangelsdorf, D.J.; Meyer, U.A. Pregnane X receptor is a target of farnesoid X receptor. J. Biol. Chem. 2006, 281, 19081–19091. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharm. Genom. 2010, 20, 1–8. [Google Scholar] [CrossRef]
- Haughton, E.L.; Tucker, S.J.; Marek, C.J.; Durward, E.; Leel, V.; Bascal, Z.; Monaghan, T.; Koruth, M.; Collie-Duguid, E.; Mann, D.A.; et al. Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology 2006, 131, 194–209. [Google Scholar] [CrossRef]
- Marek, C.J.; Tucker, S.J.; Konstantinou, D.K.; Elrick, L.J.; Haefner, D.; Sigalas, C.; Murray, G.I.; Goodwin, B.; Wright, M.C. Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem. J. 2005, 387, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Shi, J.J.; Wu, F.P.; Li, M.; Zhang, X.; Li, Y.P.; Zhai, S.; Jia, X.L.; Dang, S.S. Caffeic acid phenethyl ester up-regulates antioxidant levels in hepatic stellate cell line T6 via an Nrf2-mediated mitogen activated protein kinases pathway. World J. Gastroenterol. 2017, 23, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Kim, K.M.; Cho, S.S.; Shin, S.M.; Ka, S.O.; Na, C.S.; Park, B.H.; Jegal, K.H.; Kim, J.K.; Ku, S.K.; et al. Inhibitory Effect of Sestrin 2 on Hepatic Stellate Cell Activation and Liver Fibrosis. Antioxid. Redox. Signal. 2019, 31, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Fayez, A.M.; Zakaria, S.; Moustafa, D. Alpha lipoic acid exerts antioxidant effect via Nrf2/HO-1 pathway activation and suppresses hepatic stellate cells activation induced by methotrexate in rats. Biomed. Pharmacother. 2018, 105, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y. Curcumin targets multiple pathways to halt hepatic stellate cell activation: Updated mechanisms in vitro and in vivo. Dig. Dis. Sci. 2015, 60, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kim, S.; Hwang, S.; Cherrington, N.J.; Ryu, D.Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 2017, 8, 63370–63381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández–Gea, V.; Ghiassi–Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy Releases Lipid That Promotes Fibrogenesis by Activated Hepatic Stellate Cells in Mice and in Human Tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Shirakami, Y.; Lee, S.A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 2012, 1821, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Jophlin, L.L.; Koutalos, Y.; Chen, C.; Shah, V.; Rockey, D.C. Hepatic stellate cells retain retinoid-laden lipid droplets after cellular transdifferentiation into activated myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G713–G721. [Google Scholar] [CrossRef]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.Y.; Dapito, D.H.; Pradere, J.P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef]
- Ye, Y.; Dan, Z. All-trans retinoic acid diminishes collagen production in a hepatic stellate cell line via suppression of active protein-1 and c-Jun N-terminal kinase signal. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2010, 30, 726–733. [Google Scholar] [CrossRef]
- Senoo, H.; Wake, K. Suppression of experimental hepatic fibrosis by administration of vitamin A. Lab. Invest. 1985, 52, 182–194. [Google Scholar]
- Hisamori, S.; Tabata, C.; Kadokawa, Y.; Okoshi, K.; Tabata, R.; Mori, A.; Nagayama, S.; Watanabe, G.; Kubo, H.; Sakai, Y. All-trans-retinoic acid ameliorates carbon tetrachloride-induced liver fibrosis in mice through modulating cytokine production. Liver Int. 2008, 28, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- De Minicis, S.; Candelaresi, C.; Agostinelli, L.; Taffetani, S.; Saccomanno, S.; Rychlicki, C.; Trozzi, L.; Marzioni, M.; Benedetti, A.; Svegliati-Baroni, G. Endoplasmic Reticulum stress induces hepatic stellate cell apoptosis and contributes to fibrosis resolution. Liver Int. 2012, 32, 1574–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.G.; Yee, H.F.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Wang, Q.; Huang, X.H. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression. Acta Pharmacol. Sin. 2006, 27, 715–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chan, C.C.; Kwon, O.S.; Liu, S.; McGhee, J.; Stimpson, S.A.; Chen, L.Z.; Harrington, W.W.; Symonds, W.T.; Rockey, D.C. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G902–G911. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Kamiya, A.; Sumiyoshi, H.; Tsuruya, K.; Kagawa, T.; Inagaki, Y. A Deactivation Factor of Fibrogenic Hepatic Stellate Cells Induces Regression of Liver Fibrosis in Mice. Hepatology 2020, 71, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair 2015, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.F.; Mak, K.M.; Rackovsky, O.; Lin, Y.L.; Kwong, A.J.; Loke, J.C.; Friedman, S.L. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J. Cell Physiol. 2010, 223, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Jarnagin, W.R.; Rockey, D.C.; Koteliansky, V.E.; Wang, S.S.; Bissell, D.M. Expression of variant fibronectins in wound healing: Cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J. Cell Biol. 1994, 127, 2037–2048. [Google Scholar] [CrossRef]
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In Hepatic Fibrosis, Liver Sinusoidal Endothelial Cells Acquire Enhanced Immunogenicity. J. Immunol. 2010, 185, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927. e916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papageorgiou, M.V.; Hadziyannis, E.; Tiniakos, D.; Georgiou, A.; Margariti, A.; Kostas, A.; Papatheodoridis, G.V. Serum levels of vascular endothelial growth factor in non-alcoholic fatty liver disease. Ann. Gastroenterol. 2017, 30, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Tveden-Nyborg, P. Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact. Biomedicines 2021, 9, 93. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.J.; Velazquez, V.M.; Brigstock, D.R. Fibrogenic Signaling Is Suppressed in Hepatic Stellate Cells through Targeting of Connective Tissue Growth Factor (CCN2) by Cellular or Exosomal MicroRNA-199a-5p. Am. J. Pathol. 2016, 186, 2921–2933. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, R.; Kemper, S.; Brigstock, D.R. Pathways of production and delivery of hepatocyte exosomes. J. Cell Commun. Signal 2018, 12, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, R.; Kemper, S.; Cong, M.; You, H.; Brigstock, D.R. Therapeutic effects of serum extracellular vesicles in liver fibrosis. J. Extracell Vesicles 2018, 7, 1461505. [Google Scholar] [CrossRef]
- Oakley, F.; Meso, M.; Iredale, J.P.; Green, K.; Marek, C.J.; Zhou, X.; May, M.J.; Millward-Sadler, H.; Wright, M.C.; Mann, D.A. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 2005, 128, 108–120. [Google Scholar] [CrossRef]
- Tao, L.L.; Cheng, Y.Y.; Ding, D.; Mei, S.; Xu, J.W.; Yu, J.; Ou-Yang, Q.; Deng, L.; Chen, Q.; Li, Q.Q.; et al. C/EBP-α ameliorates CCl(4)-induced liver fibrosis in mice through promoting apoptosis of hepatic stellate cells with little apoptotic effect on hepatocytes in vitro and in vivo. Apoptosis 2012, 17, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.D.; Otano, I.; Rombouts, K.; Singh, K.P.; Peppa, D.; Gill, U.S.; Böttcher, K.; Kennedy, P.T.F.; Oben, J.; Pinzani, M.; et al. TRAIL regulatory receptors constrain human hepatic stellate cell apoptosis. Sci. Rep. 2017, 7, 5514. [Google Scholar] [CrossRef]
- Guo, Z.; Li, D.; Peng, H.; Kang, J.; Jiang, X.; Xie, X.; Sun, D.; Jiang, H. Specific hepatic stellate cell-penetrating peptide targeted delivery of a KLA peptide reduces collagen accumulation by inducing apoptosis. J. Drug Target 2017, 25, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.; Abdelmalek, M.F.; Younossi, Z.M.; Yuan, J.; Pecoraro, M.L.; Seyedkazemi, S.; Fischer, L.; Bedossa, P.; et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp. Clin. Trials 2020, 89, 105922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, A.J.; Fuchs, B.C.; Masia, R.; Holmes, J.A.; Salloum, S.; Sojoodi, M.; Ferreira, D.S.; Rutledge, S.M.; Caravan, P.; Alatrakchi, N.; et al. Prolonged cenicriviroc therapy reduces hepatic fibrosis despite steatohepatitis in a diet-induced mouse model of nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 529–545. [Google Scholar] [CrossRef]
- Hayakawa, R.; Hayakawa, T.; Takeda, K.; Ichijo, H. Therapeutic targets in the ASK1-dependent stress signaling pathways. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 434–453. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.; Bedossa, P.; Ratziu, V.; Anstee, Q.; Bugianesi, E.; Sanyal, A.; Loomba, R.; Harrison, S.A.; Balabanska, R.I.; Mateva, L. The panPPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic nash: Results of the NATIVE phase 2b trial. Hepatology 2020, 72, 9A–11A. [Google Scholar]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e571. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Drug | Mode of Action | Status | Outcome | Trial No. |
|---|---|---|---|---|
| Cenicriviroc | C-C chemokine receptor type 2 and 5 dual antagonist | Phase III trial terminated due to lack of efficacy in fibrosis improvement (primary endpoint) | No results available | NCT03028740 |
| Selonsertib | Apoptosis signal-regulating kinase 1 inhibitor | Phase III trial terminated due to lack of efficacy in fibrosis improvement (primary endpoint) | ≥1-stage fibrosis improvement without worsening of NASH in 9.6% (18 mg of drug), 12.1% (6 mg of drug) and 13.2% (placebo); p-value 0.49 (18 mg) and 0.93 (6 mg) | NCT03053050 |
| Phase III trial terminated due to lack of efficacy in fibrosis improvement (primary endpoint) | ≥1-stage fibrosis improvement without worsening of NASH in 14.4% (18 mg of drug), 12.8% (6 mg of drug) and 12.8% (placebo); p-value 0.56 (18 mg) and 0.93 (6 mg) | NCT03053063 | ||
| Pioglitazone | PPARγ agonist | No improvement of fibrosis regression in phase III trial (secondary endpoint) | Decrease in fibrosis score in 44.3% (drug) and 30.6% (placebo); p-value 0.12 | NCT00063622 |
| No improvement of fibrosis regression in phase IV trial (secondary endpoint) | Decrease in fibrosis score in 46% (drug) and 33% (placebo); p-value 0.08 | NCT00227110 | ||
| Elafibranor | PPARα and PPARβ/δ dual agonist | Phase III trial did not meet the predefined primary efficacy endpoint of NASH resolution without fibrosis worsening 1 | No significant difference in the improvement of fibrosis between treatment and placebo groups | NCT02704403 |
| Lanifibranor | PPARα, PPARγ and PPARβ/δ pan-agonist | Phase IIb trial achieved NASH resolution and fibrosis regression (secondary endpoints) | ≥1-stage fibrosis improvement without worsening of NASH in 42% (1200 mg of drug), 28% (800 mg of drug) and 24% (placebo) of ITT population; p-value 0.011 (1200 mg) and 0.53 (800 mg) Resolution of NASH and fibrosis improvement in 31% (1200 mg of drug), 21% (800 mg of drug) and 7% (placebo) of ITT population; p-value <0.001 (1200 mg) and 0.017 (800 mg) | NCT03008070 |
| Obeticholic acid | FXR agonist | Ongoing phase III trial, fibrosis improvement at planned interim analysis (primary endpoint) 2 | ≥1-stage fibrosis improvement without worsening of NASH in 23% (25 mg of drug), 18% (10 mg of drug) and 12% (placebo); p-value 0.0002 (25 mg) and 0.045 (10 mg) | NCT02548351 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zisser, A.; Ipsen, D.H.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. https://doi.org/10.3390/biomedicines9040365
Zisser A, Ipsen DH, Tveden-Nyborg P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines. 2021; 9(4):365. https://doi.org/10.3390/biomedicines9040365
Chicago/Turabian StyleZisser, Alexandra, David H. Ipsen, and Pernille Tveden-Nyborg. 2021. "Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets?" Biomedicines 9, no. 4: 365. https://doi.org/10.3390/biomedicines9040365
APA StyleZisser, A., Ipsen, D. H., & Tveden-Nyborg, P. (2021). Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines, 9(4), 365. https://doi.org/10.3390/biomedicines9040365