
Pituitary Adenomas: From Diagnosis to Therapeutics

Abstract
1. Introduction
2. Epidemiology
3. Classification of Pituitary Adenomas, Their Characteristics, and Treatment Strategies
4. Prolactin-Secreting Pituitary Adenoma (Prolactinomas)
4.1. Clinical Presentation
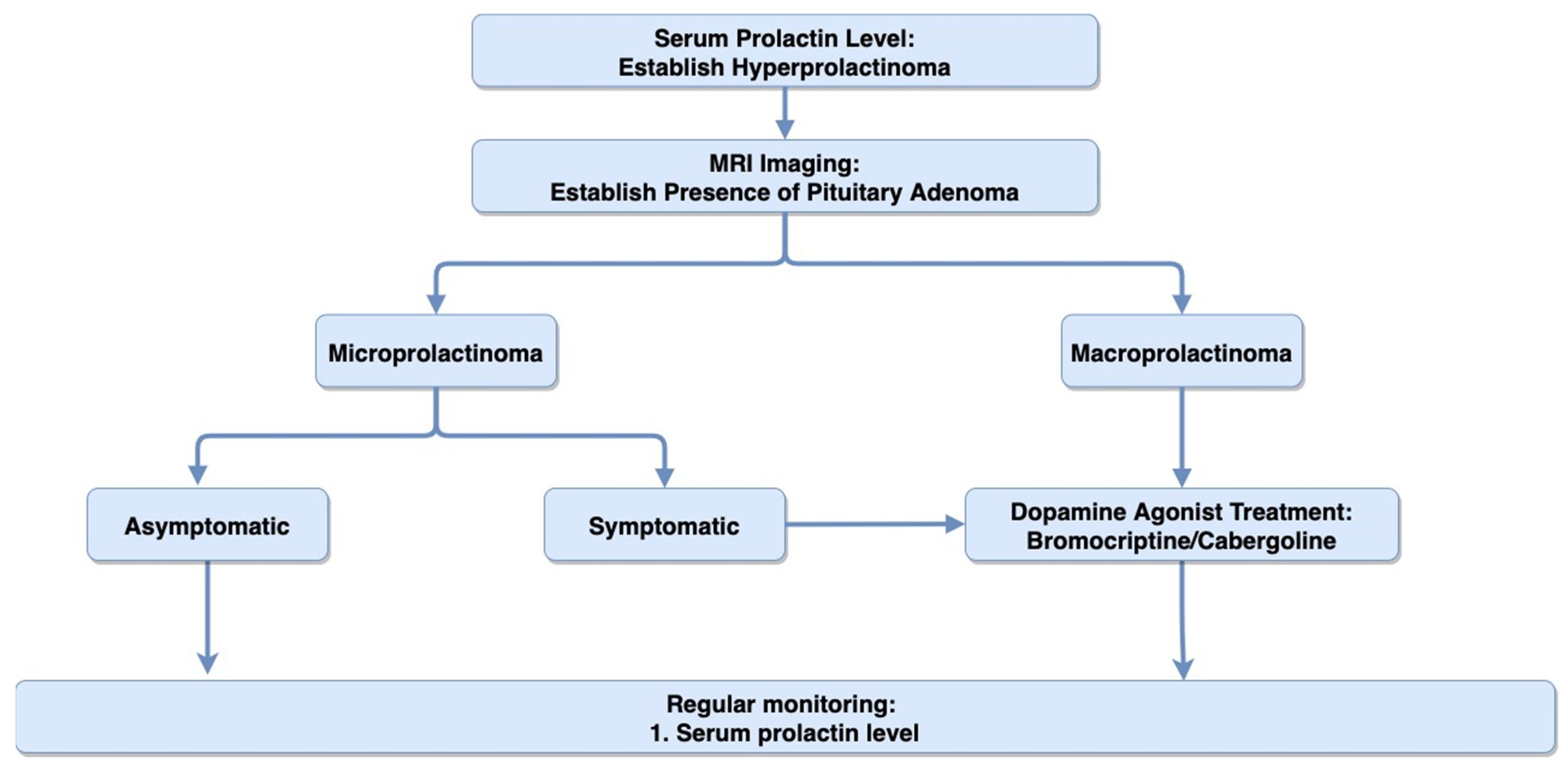
4.2. Diagnosis
4.3. Treatment
5. Acromegaly (Growth Hormone-Secreting Pituitary Adenoma)
5.1. Clinical Presentation
5.2. Diagnosis
5.3. Treatment
6. Cushing’s Syndrome (ACTH-Secreting Pituitary Adenoma)
6.1. Clinical Presentation
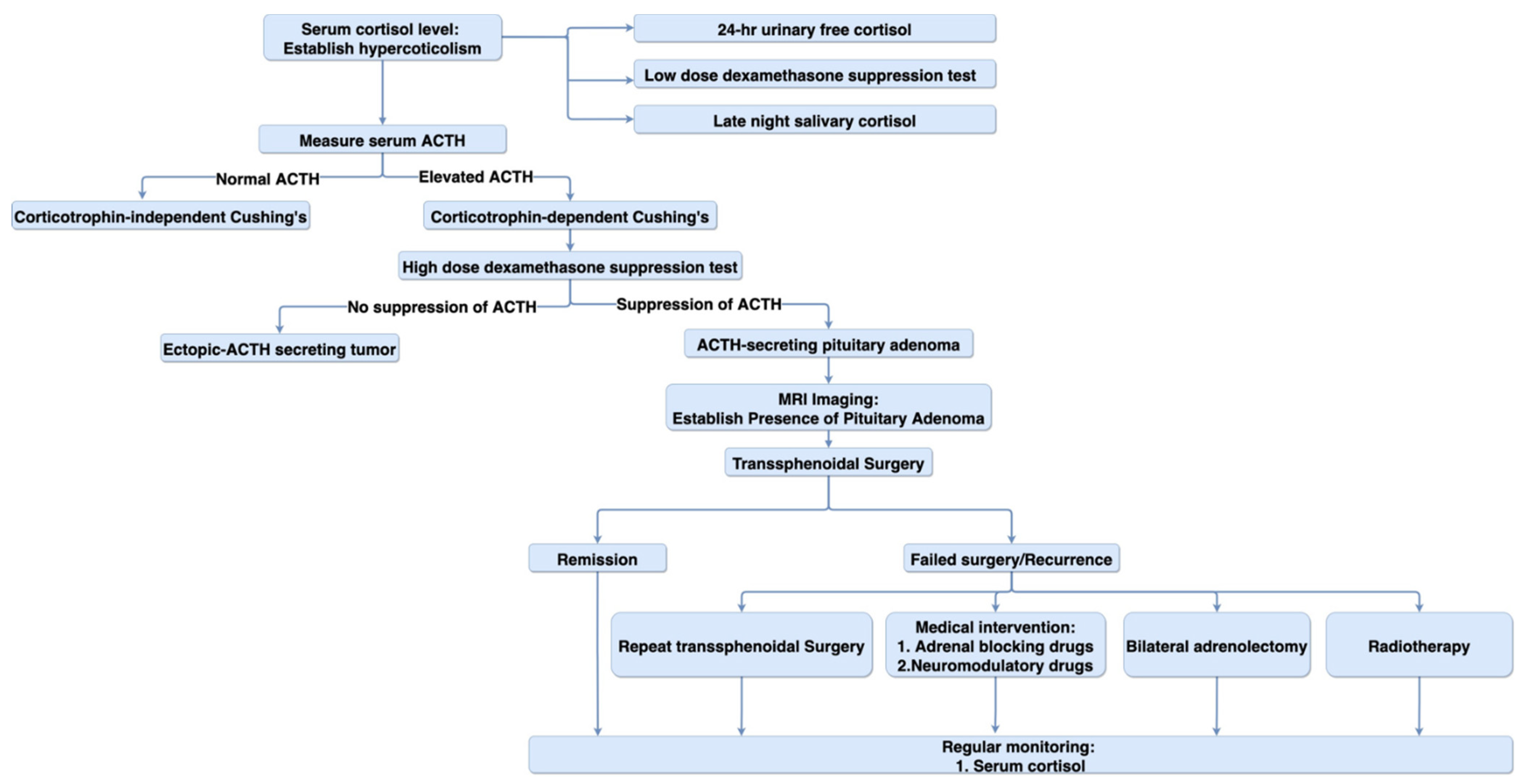
6.2. Diagnosis
6.3. Treatment
7. Non-Functioning Pituitary Adenoma
7.1. Clinical Presentation
7.2. Diagnosis
7.3. Treatment
8. Rare Pituitary Disorders
9. Future Direction
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yeh, P.J.; Chen, J.W. Pituitary tumors: Surgical and medical management. Surg. Oncol. 1997, 6, 67–92. [Google Scholar] [CrossRef]
- Dorton, A.M. The pituitary gland: Embryology, physiology, and pathophysiology. Neonatal Netw. 2000, 19, 9–17. [Google Scholar] [CrossRef]
- Melmed, S. The Pituitary; Academic Press: Cambridge, MA, USA, 2011; ISBN 9780123809261. [Google Scholar]
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The prevalence of pituitary adenomas. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lake, M.G.; Krook, L.S.; Cruz, S.V. Pituitary Adenomas: An Overview. Am. Fam. Physician 2013, 88, 319–327. [Google Scholar] [PubMed]
- Aflorei, E.D.; Korbonits, M. Epidemiology and etiopathogenesis of pituitary adenomas. J. Neuro-Oncol. 2014, 117, 379–394. [Google Scholar] [CrossRef] [PubMed]
- McNeill, K.A. Epidemiology of Brain Tumors. Neurol. Clin. 2016, 34, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Agustsson, T.T.; Baldvinsdottir, T.; Jonasson, J.G.; Olafsdottir, E.; Steinthorsdottir, V.; Sigurdsson, G.; Thorsson, A.V.; Carroll, P.V.; Korbonits, M.; Benediktsson, R. The epidemiology of pituitary adenomas in Iceland, 1955–2012: A nationwide population-based study. Eur. J. Endocrinol. 2015, 173, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Gruppetta, M.; Mercieca, C.; Vassallo, J. Prevalence and incidence of pituitary adenomas: A population based study in Malta. Pituitary 2013, 16, 545–553. [Google Scholar] [CrossRef]
- Daly, A.F.; Rixhon, M.; Adam, C.; Dempegioti, A.; Tichomirowa, M.A.; Beckers, A. High prevalence of pituitary adenomas: A cross-sectional study in the province of Liège, Belgium. J. Clin. Endocrinol. Metab. 2006, 91, 4769–4775. [Google Scholar] [CrossRef]
- Fernandez, A.; Karavitaki, N.; Wass, J.A.H. Prevalence of pituitary adenomas: A community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin. Endocrinol. (Oxf.) 2010, 72, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Raappana, A.; Koivukangas, J.; Ebeling, T.; Pirilä, T. Incidence of pituitary adenomas in Northern Finland in 1992–2007. J. Clin. Endocrinol. Metab. 2010, 95, 4268–4275. [Google Scholar] [CrossRef]
- Hall, W.A.; Luciano, M.G.; Doppman, J.L.; Patronas, N.J.; Oldfield, E.H. Pituitary magnetic resonance imaging in normal human volunteers: Occult adenomas in the general population. Ann. Intern. Med. 1994, 120, 817–820. [Google Scholar] [CrossRef]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef]
- Al-Chalabi, M.; Alsalman, I. Physiology, Prolactin; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Vroonen, L.; Daly, A.F.; Beckers, A. Epidemiology and Management Challenges in Prolactinomas. Neuroendocrinology 2019, 109, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; di Sarno, A.; Pivonello, R.; di Somma, C.; Lombardi, G. Dopamine receptor agonists for treating prolactinomas. Expert Opin. Investig. Drugs 2002, 11, 787–800. [Google Scholar] [CrossRef]
- Romijn, J.A. Hyperprolactinemia and prolactinoma. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2014; Volume 124, pp. 185–195. [Google Scholar]
- Casanueva, F.F.; Molitch, M.E.; Schlechte, J.A.; Abs, R.; Bonert, V.; Bronstein, M.D.; Brue, T.; Cappabianca, P.; Colao, A.; Fahlbusch, R.; et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin. Endocrinol. (Oxf.) 2006, 65, 265–273. [Google Scholar] [CrossRef]
- Grattan, D.R.; Jasoni, C.L.; Liu, X.; Anderson, G.M.; Herbison, A.E. Prolactin Regulation of Gonadotropin-Releasing Hormone Neurons to Suppress Luteinizing Hormone Secretion in Mice. Endocrinology 2007, 148, 4344–4351. [Google Scholar] [CrossRef]
- Koike, K.; Miyake, A.; Aono, T.; Sakamoto, T.; Ohmichi, M.; Yamaguchi, M.; Tanizaw, O. Effect of Prolactin on the Secretion of Hypothalamic GnRH and Pituitary Gonadotropins. Horm. Res. Paediatr. 1991, 35, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Deshpande, S.; Balasinor, N.H. Unveiling the Role of Prolactin and its Receptor in Male Reproduction. Horm. Metab. Res. 2019, 51, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Buvat, J. Hyperprolactinemia and sexual function in men: A short review. Int. J. Impot. Res. 2003, 15, 373–377. [Google Scholar] [CrossRef]
- Di Somma, C.; Colao, A.; Di Sarno, A.; Klain, M.; Landi, M.L.; Facciolli, G.; Pivonello, R.; Panza, N.; Salvatore, M.; Lombardi, G. Bone marker and bone density responses to dopamine agonist therapy in hyperprolactinemic males. J. Clin. Endocrinol. Metab. 1998, 83, 807–813. [Google Scholar] [CrossRef]
- Chahal, J.; Schlechte, J. Hyperprolactinemia. Pituitary 2008, 11, 141–146. [Google Scholar] [CrossRef]
- Capozzi, A.; Scambia, G.; Pontecorvi, A.; Lello, S. Hyperprolactinemia: Pathophysiology and therapeutic approach. Gynecol. Endocrinol. 2015, 31, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Arafah, B.M.; Prunty, D.; Ybarra, J.; Hlavin, M.L.; Selman, W.R. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J. Clin. Endocrinol. Metab. 2000, 85, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Klibanski, A. Prolactinomas. N. Engl. J. Med. 2010, 362, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Eloy, J.A.; Couldwell, W.T.; Liu, J.K. Update on prolactinomas. Part 1: Clinical manifestations and diagnostic challenges. J. Clin. Neurosci. 2015, 22, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Schlechte, J.A. Prolactinoma. N. Engl. J. Med. 2003, 349, 2035–2041. [Google Scholar] [CrossRef]
- Melmed, S.; Casanueva, F.F.; Hoffman, A.R.; Kleinberg, D.L.; Montori, V.M.; Schlechte, J.A.; Wass, J.A.H. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline Method of Development of Evidence-Based Clinical Practice Guidelines. J. Clin. Endocrinol. Metab. 2011, 96, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Gillam, M.P.; Molitch, M.E.; Lombardi, G.; Colao, A. Advances in the treatment of prolactinomas. Endocr. Rev. 2006, 27, 485–534. [Google Scholar] [CrossRef]
- Molitch, M.E. Diagnosis and treatment of pituitary adenomas: A review. JAMA J. Am. Med. Assoc. 2017, 317, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Sun, R.; Wen, G.; Zhong, C.; Yang, J.; Zhu, J.; Cong, Z.; Luo, X.; Ma, C. Bromocriptine and cabergoline induce cell death in prolactinoma cells via the ERK/EGR1 and AKT/mTOR pathway respectively. Cell Death Dis. 2019, 10, 335. [Google Scholar] [CrossRef]
- Delgrange, E.; Daems, T.; Verhelst, J.; Abs, R.; Maiter, D. Characterization of resistance to the prolactin-lowering effects of cabergoline in macroprolactinomas: A study in 122 patients. Eur. J. Endocrinol. 2009, 160, 747–752. [Google Scholar] [CrossRef]
- Colao, A.; Pivonello, R.; Di Somma, C.; Savastano, S.; Grasso, L.F.S.; Lombardi, G. Medical therapy of pituitary adenomas: Effects on tumor shrinkage. Rev. Endocr. Metab. Disord. 2009, 10, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.M.; Ferrari, C.; Baroldi, P.; Cavioni, V.; Paracchi, A.; Galparoli, C.; Romano, C.; Spellecchia, D.; Gerevini, G.; Crosignani, P.G. Prolactin-lowering effect of acute and once weekly repetitive oral administration of cabergoline at two dose levels in hyperprolactinemic patients. J. Clin. Endocrinol. Metab. 1988, 66, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.; Piscitelli, G.; Polli, A.; Ferrari, C.I.; Ismail, I.; Scanlon, M.F. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. N. Engl. J. Med. 1994, 331, 904–909. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Colao, A.; Di Sarno, A.; Ferone, D.; Landi, M.L.; Zarrilli, S.; Paesano, L.; Merola, B.; Lombardi, G. Cabergoline treatment rapidly improves gonadal function in hyperprolactinemic males: A comparison with bromocriptine Mechanisms of resistance to medical treatment in Pituitary adenomas View project Ascendis Pharma View project. Eur. J. Endocrinol. 1998, 138, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Di Sarno, A.; Sarnacchiaro, F.; Ferone, D.; Di Renzo, G.; Merola, B.; Annunziato, L.; Lombardi, G. Prolactinomas resistant to standard dopamine agonists respond to chronic cabergoline treatment. J. Clin. Endocrinol. Metab. 1997, 82, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Delgrange, E.; Maiter, D.; Donckier, J. Effects of the dopamine agonist cabergoline in patients with prolactinoma intolerant or resistant to bromocriptine. Eur. J. Endocrinol. 1996, 134, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Zhai, W.; Tang, H.; Hui, G.Z.; Wu, Z.B. Cabergoline for the treatment of bromocriptine-resistant invasive giant prolactinomas. Endocrine 2018, 62, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E. Management of medically refractory prolactinoma. J. Neurooncol. 2014, 117, 421–428. [Google Scholar] [CrossRef]
- Wang, A.T.; Mullan, R.J.; Lane, M.A.; Hazem, A.; Prasad, C.; Gathaiya, N.W.; Fernández-Balsells, M.M.; Bagatto, A.; Coto-Yglesias, F.; Carey, J.; et al. Treatment of hyperprolactinemia: A systematic review and meta-analysis. Syst. Rev. 2012, 1, 33. [Google Scholar] [CrossRef]
- Zygourakis, C.C.; Imber, B.S.; Chen, R.; Han, S.J.; Blevins, L.; Molinaro, A.; Kahn, J.G.; Aghi, M.K. Cost-Effectiveness Analysis of Surgical versus Medical Treatment of Prolactinomas. J. Neurol. Surg. Part B Skull Base 2017, 78, 125–131. [Google Scholar] [CrossRef]
- Chanson, P.; Maiter, D. The epidemiology, diagnosis and treatment of Prolactinomas: The old and the new. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101290. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E.; Drummond, J.; Korbonits, M. Prolactinoma Management. 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279174/ (accessed on 24 January 2021).
- Schade, R.; Andersohn, F.; Suissa, S.; Haverkamp, W.; Garbe, E. Dopamine Agonists and the Risk of Cardiac-Valve Regurgitation. N. Engl. J. Med. 2007, 356, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zanettini, R.; Antonini, A.; Gatto, G.; Gentile, R.; Tesei, S.; Pezzoli, G. Valvular Heart Disease and the Use of Dopamine Agonists for Parkinson’s Disease. N. Engl. J. Med. 2007, 356, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Delgado, V.; Biermasz, N.R.; Van Thiel, S.W.; Ewe, S.H.; Marsan, N.A.; Holman, E.R.; Feelders, R.A.; Smit, J.W.A.; Bax, J.J.; Pereira, A.M. Changes in heart valve structure and function in patients treated with dopamine agonists for prolactinomas, a 2-year follow-up study. Clin. Endocrinol. (Oxf.) 2012, 77, 99–105. [Google Scholar] [CrossRef]
- Kars, M.; Delgado, V.; Holman, E.R.; Feelders, R.A.; Smit, J.W.A.; Romijn, J.A.; Bax, J.J.; Pereira, A.M. Aortic valve calcification and mild tricuspid regurgitation but no clinical heart disease after 8 years of dopamine agonist therapy for prolactinoma. J. Clin. Endocrinol. Metab. 2008, 93, 3348–3356. [Google Scholar] [CrossRef] [PubMed]
- Drake, W.M.; Stiles, C.E.; Howlett, T.A.; Toogood, A.A.; Bevan, J.S.; Steeds, R.P. A cross-sectional study of the prevalence of cardiac valvular abnormalities in hyperprolactinemic patients treated with ergot-derived dopamine agonists. J. Clin. Endocrinol. Metab. 2014, 99, 90–96. [Google Scholar] [CrossRef]
- Valassi, E.; Klibanski, A.; Biller, B.M.K. Potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J. Clin. Endocrinol. Metab. 2010, 95, 1025–1033. [Google Scholar] [CrossRef]
- Society for Endocrinology. Position Statement on the Use of Dopamine Agonists in Endocrine Disorders; Society for Endocrinology: Bristol, UK, 2011. [Google Scholar]
- Schlechte, J.A. Long-Term Management of Prolactinomas. J. Clin. Endocrinol. Metab. 2007, 92, 2861–2865. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Di Sarno, A.; Cappabianca, P.; Di Somma, C.; Pivonello, R.; Lombardi, G. Withdrawal of Long-Term Cabergoline Therapy for Tumoral and Nontumoral Hyperprolactinemia. N. Engl. J. Med. 2003, 349, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Smith, J.; Jadon, D.; McEwan, P.; Rees, D.A.; Evans, L.M.; Scanlon, M.F.; Davies, J.S. Long-term remission following withdrawal of dopamine agonist therapy in subjects with microprolactinomas. Clin. Endocrinol. (Oxf.) 2005, 63, 26–31. [Google Scholar] [CrossRef]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Investig. 2009, 119, 3189–3202. [Google Scholar] [CrossRef]
- Katznelson, L.; Laws, E.R.; Melmed, S.; Molitch, M.E.; Murad, M.H.; Utz, A.; Wass, J.A.H. Acromegaly: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 3933–3951. [Google Scholar] [CrossRef] [PubMed]
- Vilar, L.; Vilar, C.F.; Lyra, R.; Lyra, R.; Naves, L.A. Acromegaly: Clinical features at diagnosis. Pituitary 2017, 20, 22–32. [Google Scholar] [CrossRef]
- Melmed, S. Acromegaly. N. Engl. J. Med. 2006, 355, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Le Roith, D.; Scavo, L.; Butler, A. What is the role of circulating IGF-I? Trends Endocrinol. Metab. 2001, 12, 48–52. [Google Scholar] [CrossRef]
- Lugo, G.; Pena, L.; Cordido, F. Clinical manifestations and diagnosis of acromegaly. Int. J. Endocrinol. 2012, 2012, 540398. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Ferone, D.; Marzullo, P.; Lombardi, G. Systemic Complications of Acromegaly: Epidemiology, Pathogenesis, and Management. Endocr. Rev. 2004, 25, 102–152. [Google Scholar] [CrossRef]
- Giustina, A.; Chanson, P.; Bronstein, M.D.; Klibanski, A.; Lamberts, S.; Casanueva, F.F.; Trainer, P.; Ghigo, E.; Ho, K.; Melmed, S. A consensus on criteria for cure of acromegaly. J. Clin. Endocrinol. Metab. 2010, 95, 3141–3148. [Google Scholar] [CrossRef]
- Colao, A.; Grasso, L.F.S.; Giustina, A.; Melmed, S.; Chanson, P.; Pereira, A.M.; Pivonello, R. Acromegaly. Nat. Rev. Dis. Prim. 2019, 5, 1–17. [Google Scholar] [CrossRef]
- Renehan, A.G.; O’Connell, J.; O’Halloran, D.; Shanahan, F.; Potten, C.S.; O’Dwyer, S.T.; Shalet, S.M. Acromegaly and Colorectal Cancer: A Comprehensive Review of Epidemiology, Biological Mechanisms, and Clinical Implications. Horm. Metab. Res. 2003, 35, 712–725. [Google Scholar]
- Wolinski, K.; Czarnywojtek, A.; Ruchala, M. Risk of thyroid nodular disease and thyroid cancer in patients with acromegaly—Meta-analysis and systematic review. PLoS ONE 2014, 9, e88787. [Google Scholar] [CrossRef]
- Lavrentaki, A.; Paluzzi, A.; Wass, J.A.H.; Karavitaki, N. Epidemiology of acromegaly: Review of population studies. Pituitary 2017, 20, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.J.; Post, K.D.; Bruce, J.N.; Nabi Kanibir, M.; Reyes-Vidal, C.M.; Freda, P.U. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: Acromegaly remains under-recognized and under-diagnosed. Clin. Endocrinol. (Oxf.) 2010, 72, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, D.A.; Reinholz, C.; Bauer, M.; Tufman, A.; Frohner, R.; Schopohl, J.; Bidlingmaier, M.; Kosilek, R.P.; Reincke, M.; Schneider, H.J. IGF-1-based screening reveals a low prevalence of acromegaly in patients with obstructive sleep apnea. Endocrine 2018, 60, 317–322. [Google Scholar] [CrossRef]
- Attal, P.; Chanson, P. Screening of acromegaly in adults with obstructive sleep apnea: Is it worthwhile? Endocrine 2018, 61, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Learned-Miller, E.G.; Trainer, P.; Paisley, A.; Blanz, V. Early diagnosis of acromegaly: Computers vs. clinicians. Clin. Endocrinol. (Oxf.) 2011, 75, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Gong, S.; Su, L.; Howard, N.; Kong, Y. Automatic Detection of Acromegaly From Facial Photographs Using Machine Learning Methods. EBioMedicine 2018, 27, 94–102. [Google Scholar] [CrossRef]
- Melmed, S.; Jackson, I.; Kleinberg, D.; Klibanski, A. Current Treatment Guidelines for Acromegaly 1. J. Clin. Endocrinol. Metab. 1998, 83, 2646–2652. [Google Scholar] [CrossRef]
- Melmed, S.; Bronstein, M.D.; Chanson, P.; Klibanski, A.; Casanueva, F.F.; Wass, J.A.H.; Strasburger, C.J.; Luger, A.; Clemmons, D.R.; Giustina, A. A Consensus Statement on acromegaly therapeutic outcomes. Nat. Rev. Endocrinol. 2018, 14, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Gittoes, N.J.L.; Sheppard, M.C.; Johnson, A.P.; Stewart, P.M. Outcome of surgery for acromegaly—The experience of a dedicated pituitary surgeon. QJM Mon. J. Assoc. Physicians 1999, 92, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Elsheikh, M.; Stratton, I.M.; Page, R.C.L.; Adams, C.B.T.; Wass, J.A.H. Outcome of transphenoidal surgery for acromegaly and its relationship to surgical experience. Clin. Endocrinol. (Oxf.) 1999, 50, 561–567. [Google Scholar] [CrossRef]
- Shimon, L.; Cohen, Z.R.; Ram, Z.; Hadani, M. Transsphenoidal surgery for acromegaly: Endocrinological follow-up of 98 patients. Neurosurgery 2001, 48, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, X.; Yan, Y.; Qian, J.; Lu, Y.; Luo, C. Preoperative somatostatin analogs treatment in acromegalic patients with macroadenomas. A meta-analysis. Brain Dev. 2015, 37, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zhu, H.; Xing, B.; Gu, F. Prolonged preoperative treatment of acromegaly with Somatostatin analogs may improve surgical outcome in patients with invasive pituitary macroadenoma (Knosp grades 1-3): A retrospective cohort study conducted at a single center. BMC Endocr. Disord. 2017, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Lüdecke, D.K. Recent results of secondary transnasal surgery for residual or recurring acromegaly. Neurosurgery 1998, 42, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; McKean, E.L.; Barkan, A.L.; Chandler, W.F.; Sullivan, S.E. Repeat endoscopic transsphenoidal surgery for acromegaly: Remission and complications. Pituitary 2013, 16, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Luedecke, D.K.; Abe, T. Effectiveness of Secondary Transnasal Surgery in GH-Secreting Pituitary Macroadenomas. Endocr. J. 2003, 50, 635–642. [Google Scholar] [CrossRef]
- Yamada, S.; Fukuhara, N.; Oyama, K.; Takeshita, A.; Takeuchi, Y. Repeat transsphenoidal surgery for the treatment of remaining or recurring pituitary tumors in acromegaly. Neurosurgery 2010, 67, 949–956. [Google Scholar] [CrossRef]
- Nomikos, P.; Buchfelder, M.; Fahlbusch, R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure”. Eur. J. Endocrinol. 2005, 152, 379–387. [Google Scholar] [CrossRef]
- Carmichael, J.D.; Bonert, V.S.; Nuño, M.; Ly, D.; Melmed, S. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: A meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 1825–1833. [Google Scholar] [CrossRef]
- Colao, A.; Auriemma, R.S.; Pivonello, R.; Kasuki, L.; Gadelha, M.R. Interpreting biochemical control response rates with first-generation somatostatin analogues in acromegaly. Pituitary 2016, 19, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.S.; Shih, H.A. Radiation Therapy in the Management of Pituitary Adenomas. J. Clin. Endocrinol. Metab. 1992, 96, 1992–2003. [Google Scholar] [CrossRef]
- Gheorghiu, M.L.; Fleseriu, M. Stereotactic radiation therapy in pituitary adenomas, is it better than conventional radiation therapy? Acta Endocrinol. (Copenh.) 2017, 13, 476–490. [Google Scholar] [CrossRef]
- Elhateer, H.; Muanza, T.; Roberge, D.; Ruo, R.; Eldebawy, E.; Lambert, C.; Patrocinio, H.; Shenouda, G.; Souhami, L. Fractionated stereotactic radiotherapy in the treatment of pituitary macroadenomas. Curr. Oncol. 2008, 15, 32–38. [Google Scholar] [CrossRef][Green Version]
- Ding, D.; Mehta, G.U.; Patibandla, M.R.; Lee, C.C.; Liscak, R.; Kano, H.; Pai, F.Y.; Kosak, M.; Sisterson, N.D.; Martinez-Alvarez, R.; et al. Stereotactic Radiosurgery for Acromegaly: An International Multicenter Retrospective Cohort Study. Clin. Neurosurg. 2019, 84, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Abu Dabrh, A.M.; Asi, N.; Farah, W.H.; Mohammed, K.; Wang, Z.; Farah, M.H.; Prokop, L.J.; Katznelson, L.; Murad, M.H. Radiotherapy versus Radiosurgery in Treating Patients with Acromegaly: A Systematic Review and Meta-Analysis. Endocr. Pract. 2015, 21, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Isidori, A.M.; De Martino, M.C.; Newell-Price, J.; Biller, B.M.K.; Colao, A. Complications of Cushing’s syndrome: State of the art. Lancet Diabetes Endocrinol. 2016, 4, 611–629. [Google Scholar] [CrossRef]
- Orth, D.N. Cushing’s Syndrome. N. Engl. J. Med. 1995, 332, 791–803. [Google Scholar] [CrossRef]
- Bertagna, X.; Guignat, L.; Groussin, L.; Bertherat, J. Cushing’s disease. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 607–623. [Google Scholar] [CrossRef]
- Newell-Price, J.; Bertagna, X.; Grossman, A.B.; Nieman, L.K. Cushing’s syndrome. Lancet 2006, 367, 1605–1617. [Google Scholar] [CrossRef]
- Giraldi, F.P.; Moro, M.; Cavagnini, F. Gender-related differences in the presentation and course of Cushing’s disease. J. Clin. Endocrinol. Metab. 2003, 88, 1554–1558. [Google Scholar] [CrossRef]
- Pivonello, R.; De Martino, M.C.; De Leo, M.; Lombardi, G.; Colao, A. Cushing’s Syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 135–149. [Google Scholar] [CrossRef]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; Montori, V.M.; Edwards, H. The diagnosis of Cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2008, 93, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Raff, H.; Raff, J.L.; Findling, J.W. Late-night salivary cortisol as a screening test for Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1998, 83, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Raff, H. Cushing Syndrome. Update on Testing. Endocrinol. Metab. Clin. N. Am. 2015, 44, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Angeli, A.; Atkinson, A.B.; Bertagna, X.; Cavagnini, F.; Chrousos, G.P.; Fava, G.A.; Findling, J.W.; Gaillard, R.C.; Grossman, A.B.; et al. Diagnosis and Complications of Cushing’s Syndrome: A Consensus Statement. J. Clin. Endocrinol. Metab. 2003, 88, 5593–5602. [Google Scholar] [CrossRef]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Murad, M.H.; Newell-Price, J.; Savage, M.O.; Tabarin, A. Treatment of cushing’s syndrome: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2015, 100, 2807–2831. [Google Scholar] [CrossRef] [PubMed]
- Petersenn, S.; Beckers, A.; Ferone, D.; Van Der Lely, A.; Bollerslev, J.; Boscaro, M.; Brue, T.; Bruzzi, P.; Casanueva, F.F.; Chanson, P.; et al. Outcomes in patients with Cushing’s disease undergoing transsphenoidal surgery: Systematic review assessing criteria used to define remission and recurrence. Eur. J. Endocrinol. 2015, 172, R227–R239. [Google Scholar] [CrossRef]
- Donofrio, C.A.; Losa, M.; Gemma, M.; Giudice, L.; Barzaghi, L.R.; Mortini, P. Safety of transsphenoidal microsurgical approach in patients with an ACTH-secreting pituitary adenoma. Endocrine 2017, 58, 303–311. [Google Scholar] [CrossRef]
- Mampalam, T.J.; Tyrrell, J.B.; Wilson, C.B. Transsphenoidal microsurgery for Cushing disease: A report of 216 cases. Ann. Intern. Med. 1988, 109, 487–493. [Google Scholar] [CrossRef]
- Hameed, N.; Yedinak, C.G.; Brzana, J.; Gultekin, S.H.; Coppa, N.D.; Dogan, A.; Delashaw, J.B.; Fleseriu, M. Remission rate after transsphenoidal surgery in patients with pathologically confirmed Cushing’s disease, the role of cortisol, ACTH assessment and immediate reoperation: A large single center experience. Pituitary 2013, 16, 452–458. [Google Scholar] [CrossRef]
- Burke, W.T.; Penn, D.L.; Repetti, C.S.; Iuliano, S.; Laws, E.R. Outcomes after Repeat Transsphenoidal Surgery for Recurrent Cushing Disease: Updated. Clin. Neurosurg. 2019, 85, E1030–E1036. [Google Scholar] [CrossRef]
- Patil, C.G.; Veeravagu, A.; Prevedello, D.M.; Katznelson, L.; Vance, M.L.; Laws, E.R. Outcomes after repeat transsphenoidal surgery for recurrent Cushings disease. Neurosurgery 2008, 63, 266–271. [Google Scholar] [CrossRef]
- Bertagna, X.; Guignat, L. Approach to the Cushing’s Disease Patient With Persistent/Recurrent Hypercortisolism after Pituitary Surgery. J. Clin. Endocrinol. Metab. 2013, 98, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E. Current approaches to the pharmacological management of Cushing’s disease. Mol. Cell. Endocrinol. 2015, 408, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Petersenn, S.; Newell-Price, J.; Findling, J.W.; Gu, F.; Maldonado, M.; Schoenherr, U.; Mills, D.; Salgado, L.R.; Biller, B.M.; et al. A 12-Month Phase 3 Study of Pasireotide in Cushing’s Disease. N. Engl. J. Med. 2012, 366, 914–924. [Google Scholar] [CrossRef]
- Smith, P.W.; Turza, K.C.; Carter, C.O.; Vance, M.L.; Laws, E.R.; Hanks, J.B. Bilateral Adrenalectomy for Refractory Cushing Disease: A Safe and Definitive Therapy. J. Am. Coll. Surg. 2009, 208, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.K.; Hayman, A.V.; Ludlam, W.H.; Deveney, C.W.; Loriaux, D.L.; Sheppard, B.C. Improved quality of life after bilateral laparoscopic adrenalectomy for cushing’s disease: A 10-year experience. Ann. Surg. 2007, 245, 790–794. [Google Scholar] [CrossRef]
- Chow, J.T.; Thompson, G.B.; Grant, C.S.; Farley, D.R.; Richards, M.L.; Young, W.F. Bilateral laparoscopic adrenalectomy for corticotrophin-dependent Cushing’s syndrome: A review of the Mayo Clinic experience. Clin. Endocrinol. (Oxf.) 2008, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.M.; Williams, B.J.; Vance, M.L.; Sheehan, J.P. Radiation therapy and stereotactic radiosurgery for the treatment of Cushings disease: An evidence-based review. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Raverot, G.; Burman, P.; McCormack, A.; Heaney, A.; Petersenn, S.; Popovic, V.; Trouillas, J.; Dekkers, O.M. European society of endocrinology clinical practice guidelines for the management of aggressive pituitary tumours and carcinomas. Eur. J. Endocrinol. 2018, 178, G1–G24. [Google Scholar] [CrossRef]
- Freda, P.U.; Beckers, A.M.; Katznelson, L.; Molitch, M.E.; Montori, V.M.; Post, K.D.; Lee Vance, M. Pituitary incidentaloma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, C.A. Clinically non-functioning pituitary adenoma. Pituitary 2006, 9, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Shenoy, K.; Seshadri, M.S.; Muliyil, J.; Rao, A.; Paul, P. Visual field defects in non-functioning pituitary adenomas. Indian J. Ophthalmol. 2002, 50, 127. [Google Scholar]
- Abouaf, L.; Vighetto, A.; Lebas, M. Neuro-ophthalmologic exploration in non-functioning pituitary adenoma. Ann. Endocrinol. (Paris) 2015, 76, 210–219. [Google Scholar] [CrossRef]
- Ferrante, E.; Ferraroni, M.; Castrignanò, T.; Menicatti, L.; Anagni, M.; Reimondo, G.; Del Monte, P.; Bernasconi, D.; Loli, P.; Faustini-Fustini, M.; et al. Non-functioning pituitary adenoma database: A useful resource to improve the clinical management of pituitary tumors. Eur. J. Endocrinol. 2006, 155, 823–829. [Google Scholar] [CrossRef]
- Esposito, D.; Olsson, D.S.; Ragnarsson, O.; Buchfelder, M.; Skoglund, T.; Johannsson, G. Non-functioning pituitary adenomas: Indications for pituitary surgery and post-surgical management. Pituitary 2019, 22, 422–434. [Google Scholar] [CrossRef]
- Ntali, G.; Wass, J.A. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary 2018, 21, 111–118. [Google Scholar] [CrossRef]
- Chanson, P.; Raverot, G.; Castinetti, F.; Cortet-Rudelli, C.; Galland, F.; Salenave, S.; Cazabat, L.; Foubert, L.; Bonneville, J.F.; Gaillard, S.; et al. Management of clinically non-functioning pituitary adenoma. Ann. Endocrinol. (Paris) 2015, 76, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Dufour, H.; Gaillard, S.; Jouanneau, E.; Vasiljevic, A.; Villa, C.; Trouillas, J. Non-functioning pituitary adenoma: When and how to operate? What pathologic criteria for typing? Ann. Endocrinol. (Paris) 2015, 76, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, O.M.; Pereira, A.M.; Romijn, J.A. Treatment and follow-up of clinically nonfunctioning pituitary macroadenomas. J. Clin. Endocrinol. Metab. 2008, 93, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Park, P.; Chandler, W.F.; Barkan, A.L.; Orrego, J.J.; Cowan, J.A.; Griffith, K.A.; Tsien, C. The role of radiation therapy after surgical resection of nonfunctional pituitary macroadenomas. Neurosurgery 2004, 55, 100–107. [Google Scholar] [CrossRef]
- Brada, M.; Rajan, B.; Traish, D.; Ashley$, S.; Holmes-Sellorstj, P.J.; Nusseylt, S.; Uttleyll, D. The long-term efficacy of conservative surgery and radiotherapy in the control of pituitary adenomas. Clin. Endocrinol. 1993, 38, 571–578. [Google Scholar] [CrossRef]
- Tsang, R.W.; Brierley, J.D.; Panzarella, T.; Gospodarowicz, M.K.; Sutcliffe, S.B.; Simpson, W.J. Radiation therapy for pituitary adenoma: Treatment outcome and prognostic factors. Int. J. Radiat. Oncol. Biol. Phys. 1994, 30, 557–565. [Google Scholar] [CrossRef]
- Becker, G.; Kocher, M.; Kortmann, R.D.; Paulsen, F.; Jeremic, B.; Müller, R.P.; Bamberg, M. Radiation therapy in the multimodal treatment approach of pituitary adenoma. Strahlenther. Onkol. 2002, 178, 173–186. [Google Scholar] [CrossRef]
- Minniti, G.; Jaffrain-Rea, M.L.; Osti, M.; Cantore, G.; Enrici, R.M. Radiotherapy for nonfunctioning pituitary adenomas: From conventional to modern stereotactic radiation techniques. Neurosurg. Rev. 2007, 30, 167–176. [Google Scholar] [CrossRef]
- Greenman, Y.; Ouaknine, G.; Veshchev, I.; Reider-Groswasser, I.I.; Segev, Y.; Stern, N. Postoperative surveillance of clinically nonfunctioning pituitary macroadenomas: Markers of tumour quiescence and regrowth. Clin. Endocrinol. (Oxf.) 2003, 58, 763–769. [Google Scholar] [CrossRef]
- Beck-Peccoz, P.; Giavoli, C.; Lania, A. A 2019 update on TSH-secreting pituitary adenomas. J. Endocrinol. Investig. 2019, 42, 1401–1406. [Google Scholar] [CrossRef]
- Losa, M.; Fortunato, M.; Molteni, L.; Pereiti, E.; Mortini, P. Thyrotropin-secreting pituitary adenomas: Biological and molecular features, diagnosis and therapy. Minerva Endocrinol. 2008, 33, 329–340. [Google Scholar]
- Amlashi, F.G.; Tritos, N.A. Thyrotropin-secreting pituitary adenomas: Epidemiology, diagnosis, and management. Endocrine 2016, 52, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kwon, B.J.; Na, D.G.; Kim, J.H.; Han, M.H.; Chang, K.H. Pituitary adenoma, craniopharyngioma, and Rathke cleft cyst involving both intrasellar and suprasellar regions: Differentiation using MRI. Clin. Radiol. 2007, 62, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Langrand, C.; Bihan, H.; Raverot, G.; Varron, L.; Androdias, G.; Borson-Chazot, F.; Brue, T.; Cathebras, P.; Pinede, L.; Muller, G.; et al. Hypothalamo-pituitary sarcoidosis: A multicenter study of 24 patients. QJM 2012, 105, 981–995. [Google Scholar] [CrossRef]
- Johnsen, D.E.; Woodruff, W.W.; Allen, I.S.; Cera, P.J.; Funkhouser, G.R.; Coleman, L.L. MR imaging of the sellar and juxtasellar regions. Radiographics 1991, 11, 727–758. [Google Scholar] [CrossRef]
- Lakomkin, N.; Van Gompel, J.J.; Post, K.D.; Cho, S.S.; Lee, J.Y.K.; Hadjipanayis, C.G. Fluorescence guided surgery for pituitary adenomas. J. Neurooncol. 2021, 151, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Aihara, Y.; Tsuzuki, S.; Okada, Y.; Kawamata, T. Application of indocyanine green fluorescence endoscopic system in transsphenoidal surgery for pituitary tumors. Acta Neurochir. (Wien) 2019, 161, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Litvack, Z.N.; Zada, G.; Laws, E.R. Indocyanine green fluorescence endoscopy for visual differentiation of pituitary tumor from surrounding structures: Clinical article. J. Neurosurg. 2012, 116, 935–941. [Google Scholar] [CrossRef]
- Lesueur, P.; Calugaru, V.; Nauraye, C.; Stefan, D.; Cao, K.; Emery, E.; Reznik, Y.; Habrand, J.L.; Tessonnier, T.; Chaikh, A.; et al. Proton therapy for treatment of intracranial benign tumors in adults: A systematic review. Cancer Treat. Rev. 2019, 72, 56–64. [Google Scholar] [CrossRef]
- Wattson, D.A.; Tanguturi, S.K.; Spiegel, D.Y.; Niemierko, A.; Biller, B.M.K.; Nachtigall, L.B.; Bussière, M.R.; Swearingen, B.; Chapman, P.H.; Loeffler, J.S.; et al. Outcomes of proton therapy for patients with functional pituitary adenomas. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 532–539. [Google Scholar] [CrossRef]
- Shi, C.; Ye, Z.; Han, J.; Ye, X.; Lu, W.; Ji, C.; Li, Z.; Ma, Z.; Zhang, Q.; Zhang, Y.; et al. BRD4 as a therapeutic target for nonfunctioning and growth hormone pituitary adenoma. Neuro-Oncology 2020, 22, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Desiderio, D.M. Signaling pathway networks mined from human pituitary adenoma proteomics data. BMC Med. Genom. 2010, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Wu, B.; Li, J.; Wang, X.; Jiang, S.; Hu, F.; Dou, G.; Zhang, Y.; Sheng, C.; Zhao, G.; et al. T5224, RSPO2 and AZD5363 are novel drugs against functional pituitary adenoma. Aging 2019, 11, 9043–9059. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ma, C.X.; Xing, Y.Z.; Yan, Z.Y. Identification of candidate target genes of pituitary adenomas based on the DNA microarray. Mol. Med. Rep. 2016, 13, 2182–2186. [Google Scholar] [CrossRef] [PubMed][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Demographic | Collection Period | Total PA Cases Identified | Prevalence (per 100,000) | Proportion of all PA (%) | Proportion of All PA (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| PRL-PA 3 | ACTH-PA 4 | GH-PA 5 | NF-PA 6 | Macroadenoma | Microadenoma | |||||
| Agustsson et al., 1,2 [8] | Iceland | 58 years (1955–2012) | 471 (190 M, 281 F) | 116 | 39.9 | 11.3 | 5.7 | 43.1 | 54.8 | 41.2 |
| Gruppetta et al., [9] | Malta | 11 years (2000–2011) | 316 (96 M, 220 F) | 76 | 46.2 | 2.2 | 16.5 | 34.2 | 43.4 | 56.6 |
| Daly et al., [10] | Belgium | Data as of 30 September 2005 | 68 (22 M, 46 F) | 94 | 66.2 | 5.9 | 13.2 | 14.7 | 42.6 | 57.3 |
| Fernandez et al., [11] | United Kingdom | Data as of 31 July 2006 | 63 (21 M, 42 F) | 78 | 57 | 2 | 11 | 28 | 41.3 | 58.7 |
| Raappana et al., [12] | Finland | 17 years (1992–2007) | 164 (47 M, 117 F) | 68 | 51 | 3 | 8.5 | 37 | 54 | 46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banskota, S.; Adamson, D.C. Pituitary Adenomas: From Diagnosis to Therapeutics. Biomedicines 2021, 9, 494. https://doi.org/10.3390/biomedicines9050494
Banskota S, Adamson DC. Pituitary Adenomas: From Diagnosis to Therapeutics. Biomedicines. 2021; 9(5):494. https://doi.org/10.3390/biomedicines9050494
Chicago/Turabian StyleBanskota, Samridhi, and David C. Adamson. 2021. "Pituitary Adenomas: From Diagnosis to Therapeutics" Biomedicines 9, no. 5: 494. https://doi.org/10.3390/biomedicines9050494
APA StyleBanskota, S., & Adamson, D. C. (2021). Pituitary Adenomas: From Diagnosis to Therapeutics. Biomedicines, 9(5), 494. https://doi.org/10.3390/biomedicines9050494