
Opposed Interplay between IDH1 Mutations and the WNT/β-Catenin Pathway: Added Information for Glioma Classification

Abstract
:1. Introduction
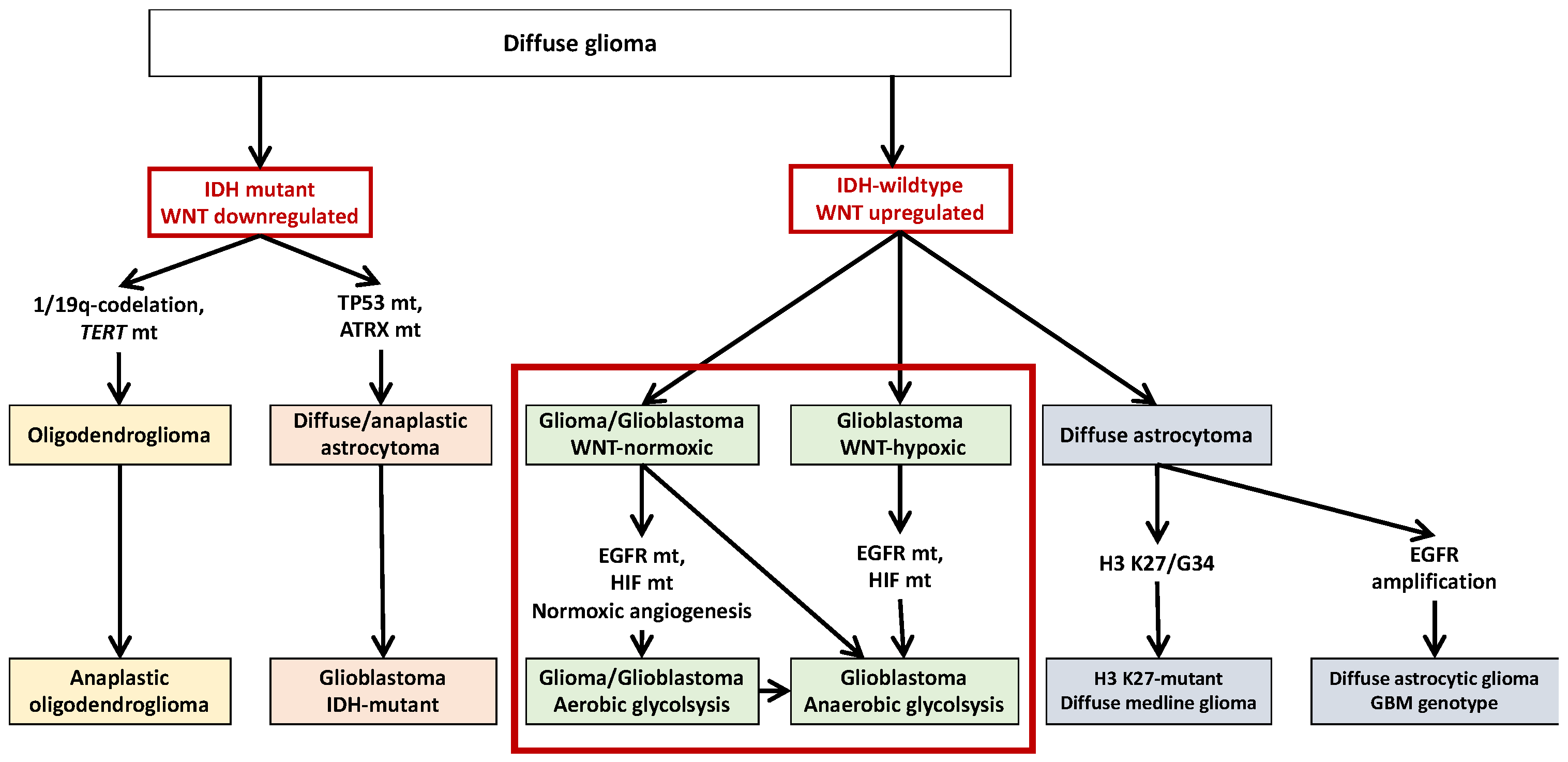
2. IDH1 Mutations and Glioma
3. Non-IDH1-Mutated Glioma
4. Canonical WNT/β-Catenin Pathway
5. Canonical WNT/β-Catenin Pathway in Glioma
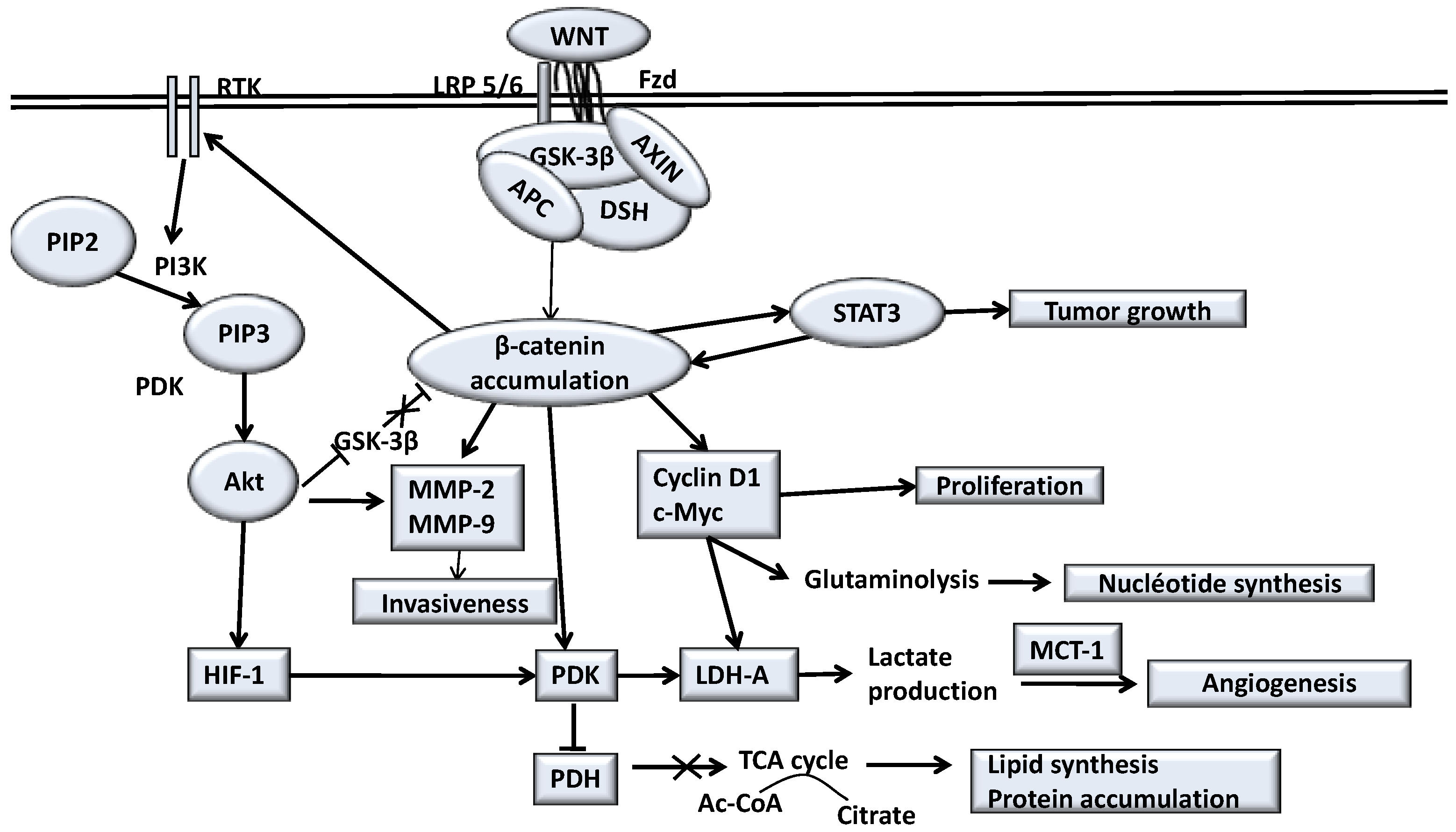
6. Opposed Interplay between IDH1 Mutations and WNT/β-Catenin Pathway in Glioma
7. Oxidative Stress and IDH1 Mutations in Glioma
8. Hypoxia in IDH1-Mutated Glioma
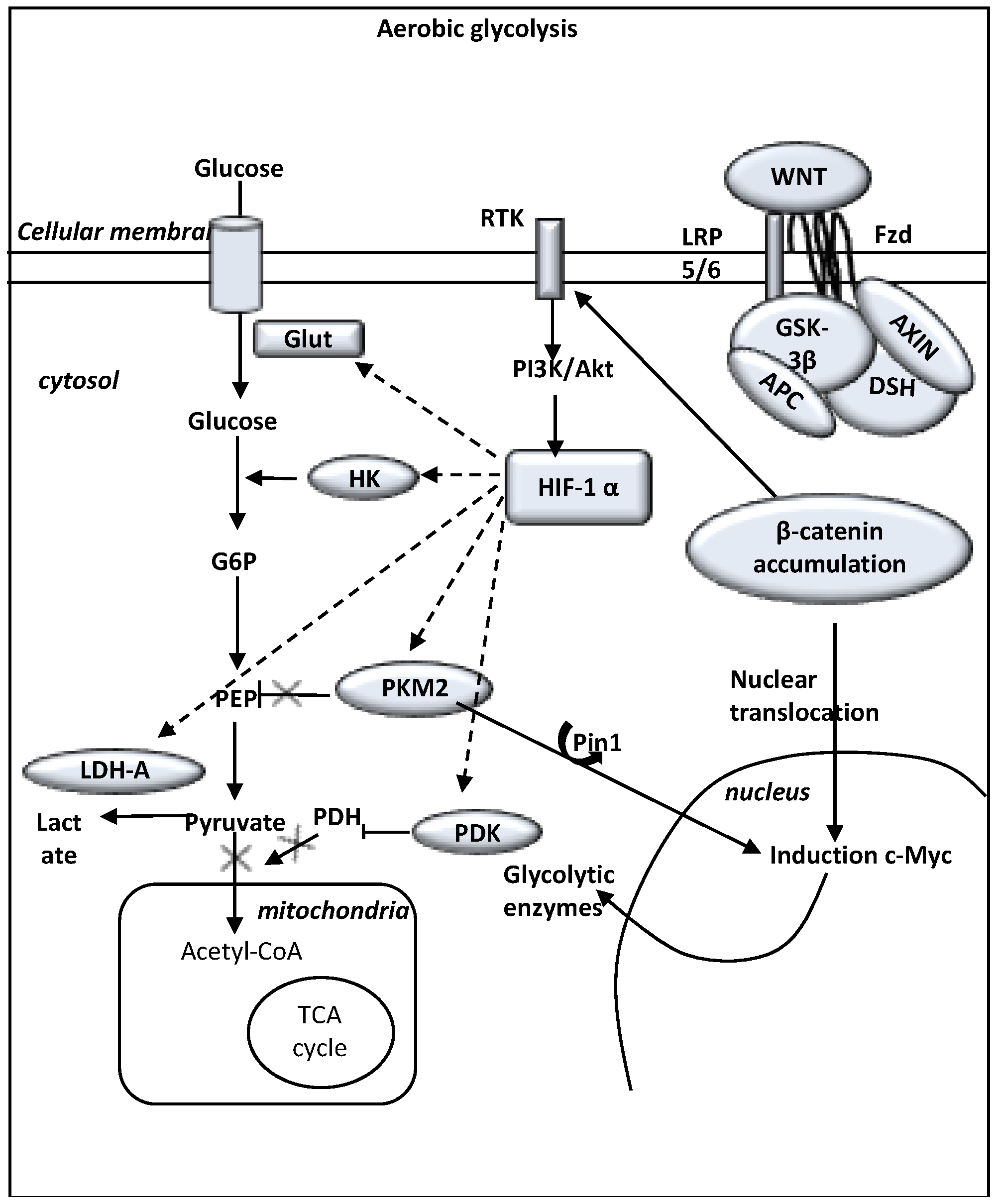
9. Aerobic Glycolysis and WNT/β-catenin Pathway in Glioma
10. Normoxia and WNT/β-catenin Pathway Upregulation in Glioma
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Gladson, C.L.; Prayson, R.A.; Liu, W. (Michael) The Pathobiology of Glioma Tumors. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, B.; Priesterbach-Ackley, L.; Petersen, J.; Wesseling, P. Molecular pathology of tumors of the central nervous system. Ann. Oncol. 2019, 30, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, H.; Narita, Y.; Yoshida, A.; Hashimoto, N.; Yoshimine, T.; Ichimura, K. IDH1/2 mutation detection in gliomas. Brain Tumor Pathol. 2014, 32, 79–89. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, F.; Chou, F.-J.; Zaghloul, K.A.; Yang, C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines 2020, 8, 294. [Google Scholar] [CrossRef]
- Ülgen, E.; Karacan, S.; Gerlevik, U.; Can, O.; Bilguvar, K.; Oktay, Y.; Akyerli, C.B.; Yüksel, Ş.K.; Danyeli, A.E.; Tihan, T.; et al. Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms. Biomedicines 2020, 8, 574. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Gong, A.; Huang, S. FoxM1 and Wnt/β-Catenin Signaling in Glioma Stem Cells. Cancer Res. 2012, 72, 5658–5662. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-Y.; Liang, G.-B.; Du, P.; Liu, Y.-H. Lgr4 Promotes Glioma Cell Proliferation through Activation of Wnt Signaling. Asian Pac. J. Cancer Prev. 2013, 14, 4907–4911. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2015, 96, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, B.; Song, Y.; Wu, Y.; Yang, X.; Peng, T.; Peng, L.; Xia, K.; Xia, X.; Chen, L.; Zhong, C. Matrix stiffness promotes glioma cell stemness by activating BCL9L/Wnt/β-catenin signaling. Aging 2021, 13, 5284–5296. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nat. Cell Biol. 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [PubMed] [Green Version]
- Agnihotri, S.; Aldape, K.D.; Zadeh, G. Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurg. Focus 2014, 37, E13. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; Von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Ducray, F.; Marie, Y.; Sanson, M. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 2248–2249, author reply 2249. [Google Scholar] [PubMed]
- Songtao, Q.; Lei, Y.; Si, G.; Yanqing, D.; Huixia, H.; Xuelin, Z.; Lanxiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Tesileanu, C.M.S.; Dirven, L.; Wijnenga, M.M.J.; Koekkoek, J.A.F.; Vincent, E.A.J.P.; Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; van Duinen, S.G.; Smits, M.; et al. Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: A confirmation of the cIMPACT-NOW criteria. Neuro-Oncology 2020, 22, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; De Biase, D.; Di Nunno, V.; Pession, A.; Tosoni, A.; Gatto, L.; Tallini, G.; Visani, M.; Lodi, R.; Bartolini, S.; et al. IDH1 Non-Canonical Mutations and Survival in Patients with Glioma. Diagnostics 2021, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neuro-Oncology 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.W.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-Oncology 2009, 11, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; Von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Weller, M.; Felsberg, J.; Hartmann, C.; Berger, H.; Steinbach, J.P.; Schramm, J.; Westphal, M.; Schackert, G.; Simon, M.; Tonn, J.C.; et al. Molecular Predictors of Progression-Free and Overall Survival in Patients with Newly Diagnosed Glioblastoma: A Prospective Translational Study of the German Glioma Network. J. Clin. Oncol. 2009, 27, 5743–5750. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Burger, P.; Kleihues, P. Definition of Primary and Secondary Glioblastoma—Response. Clin. Cancer Res. 2014, 20, 2013. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nat. Cell Biol. 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Hutter, B.; Jäger, N.; Korshunov, A.; Kool, M.; Warnatz, H.-J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.K.; et al. Recurrent Somatic Alterations of FGFR1 and NTRK2 in Pilocytic Astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nat. Cell Biol. 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Bhavya, B.; Anand, C.R.; Madhusoodanan, U.K.; Rajalakshmi, P.; Krishnakumar, K.; Easwer, H.V.; Deepti, A.N.; Gopala, S. To be wild or Mutant: Role of Isocitrate Dehydrogenase 1 (IDH1) and 2-Hydroxy Glutarate (2-HG) in Gliomagenesis and Treatment Outcome in Glioma. Cell. Mol. Neurobiol. 2019, 40, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Dubbink, H.J.; Taal, W.; Van Marion, R.; Kros, J.M.; Van Heuvel, I.; Bromberg, J.E.; Zonnenberg, B.A.; Zonnenberg, C.B.L.; Postma, T.J.; Gijtenbeek, J.M.M.; et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 2009, 73, 1792–1795. [Google Scholar] [CrossRef]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Gorovets, D.; Kannan, K.; Shen, R.; Kastenhuber, E.R.; Islamdoust, N.; Campos, C.; Pentsova, E.; Heguy, A.; Jhanwar, S.C.; Mellinghoff, I.K.; et al. IDH Mutation and Neuroglial Developmental Features Define Clinically Distinct Subclasses of Lower Grade Diffuse Astrocytic Glioma. Clin. Cancer Res. 2012, 18, 2490–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Franceschi, E.; De Biase, D.; Di Nunno, V.; Pession, A.; Tosoni, A.; Gatto, L.; Lodi, R.; Tallini, G.; Visani, M.; Bartolini, S.; et al. IDH1105GGT single nucleotide polymorphism improves progression free survival in patients with IDH mutated grade II and III gliomas. Pathol. Res. Pract. 2021, 221, 153445. [Google Scholar] [CrossRef]
- Tesileanu, C.M.S.; Vallentgoed, W.R.; Sanson, M.; Taal, W.; Clement, P.M.; Wick, W.; Brandes, A.A.; Baurain, J.F.; Chinot, O.L.; Wheeler, H.; et al. Non-IDH1-R132H IDH1/2 mutations are associated with increased DNA methylation and improved survival in astrocytomas, compared to IDH1-R132H mutations. Acta Neuropathol. 2021, 141, 945–957. [Google Scholar] [CrossRef]
- Kessler, J.; Hohmann, T.; Güttler, A.; Petrenko, M.; Ostheimer, C.; Hohmann, U.; Bache, M.; Dehghani, F.; Vordermark, D. Radiosensitization and a Less Aggressive Phenotype of Human Malignant Glioma Cells Expressing Isocitrate Dehydrogenase 1 (IDH1) Mutant Protein: Dissecting the Mechanisms. Cancers 2019, 11, 889. [Google Scholar] [CrossRef] [Green Version]
- Gülten, G.; Yalçın, N.; Baltalarlı, B.; Doğu, G.; Acar, F.; Doğruel, Y. The importance of IDH1, ATRX and WT-1 mutations in glioblastoma. Pol. J. Pathol. 2020, 71, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Weber, R.; Willscher, E.; Riehmer, V.; Hentschel, B.; Kreuz, M.; Felsberg, J.; Beyer, U.; Löffler-Wirth, H.; Kaulich, K.; et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015, 129, 679–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, M.; Barthel, F.; Malta, T.M.; Sabedot, T.S.; Salama, S.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.W.; Aldape, K.D.; Yung, W.K.A.; Salama, S.R.; Cooper, L.A.D.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR Mutation-Induced Alternative Splicing of Max Contributes to Growth of Glycolytic Tumors in Brain Cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-L.; Chang, H.-H.; Chen, Y.-C.; Chang, Y.-C.; Tsai, Y.C.A.W.-C. Molecular Mechanisms of KDELC2 on Glioblastoma Tumorigenesis and Temozolomide Resistance. Biomedicines 2020, 8, 339. [Google Scholar] [CrossRef]
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus 2015, 38, E4. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2010, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, P.K.; Hart, J. PI3K and STAT3: A New Alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.-W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumor Biol. 2013, 34, 1991–2002. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P.T. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Hamilton, R.L. Molecular diagnostics of gliomas. Arch. Pathol. Lab. Med. 2011, 135, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Mischel, P.S.; Reifenberger, G. Molecular classification of gliomas. Handb. Clin. Neurol. 2016, 134, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.A.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Clarke, I.D.; Dirks, P.B. A human brain tumor-derived PDGFR-α deletion mutant is transforming. Oncogene 2003, 22, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.S.; Wang, X.-Y.; Qian, J.; Hosek, S.M.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Amplification of the platelet-derived growth factor receptor-A (PDGFRA) gene occurs in oligodendrogliomas with grade IV anaplastic features. J. Neuropathol. Exp. Neurol. 2000, 59, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokker, A.N.; Sullivan, C.M.; Hollenbach, S.J.; Israel, A.M.; Giese, A.N. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar]
- Assanah, M.; Lochhead, R.; Ogden, A.; Bruce, J.; Goldman, J.; Canoll, P. Glial Progenitors in Adult White Matter Are Driven to Form Malignant Gliomas by Platelet-Derived Growth Factor-Expressing Retroviruses. J. Neurosci. 2006, 26, 6781–6790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. Rep. 2017, 13, 17–23. [Google Scholar] [CrossRef]
- Al-Harthi, L. Wnt/β-catenin and its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt your brain be inflamed? Yes, it Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [Green Version]
- LeCarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- LeCarpentier, Y.; Vallée, A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; LeCarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay between Canonical WNT/Beta-Catenin Pathway–PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromolecular Med. 2018, 20, 174–204. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway. Cells 2021, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identifi-cation of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome Proliferator-activated Receptor γ Activation Can Regulate β-Catenin Levels via a Proteasome-mediated and Adenomatous Polyposis Coli-independent Pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [Green Version]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt Signaling: Role in Alzheimer Disease and Schizophrenia. J. Neuroimmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Hypothesis of Opposite Interplay between the Canonical WNT/beta-catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.-M.; Zhou, F.-Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; Lima, L.D.S.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.; Scavone, C. Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/β-CATENIN signaling in rat hippocampus. Aging 2015, 7, 1094–1108. [Google Scholar] [CrossRef]
- Wu, J.-H.; Zhang, S.-H.; Gao, F.-J.; Lei, Y.; Chen, X.-Y.; Zhang, S.-J.; Sun, X.-H. RNAi screening identifies GSK3β as a regulator of DRP1 and the neuroprotection of lithium chloride against elevated pressure involved in downregulation of DRP1. Neurosci. Lett. 2013, 554, 99–104. [Google Scholar] [CrossRef]
- Russo, R.; Adornetto, A.; Cavaliere, F.; Varano, G.P.; Rusciano, D.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G.; Nucci, C. Intravitreal injection of forskolin, homotaurine, and L-carnosine affords neuroprotection to retinal ganglion cells following retinal ischemic injury. Mol. Vis. 2015, 21, 718–729. [Google Scholar]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay Between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; LeCarpentier, Y.; Vallée, J.-N. Targeting the Canonical WNT/β-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs. Cells 2019, 8, 726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Vallée, J.-N.; LeCarpentier, Y. PPARγ agonists: Potential treatment for autism spectrum disorder by inhibiting the canonical WNT/β-catenin pathway. Mol. Psychiatry 2019, 24, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. The influence of circadian rhythms and aerobic glycolysis in autism spectrum disorder. Transl. Psychiatry 2020, 10, 400. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Guillevin, R.; Lecarpentier, Y. Riluzole: A therapeutic strategy in Alzheimer’s disease by targeting the WNT/β-catenin pathway. Aging 2020, 12, 3095–3113. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N. Warburg effect hypothesis in autism Spectrum disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Sareddy, G.R.; Panigrahi, M.; Challa, S.; Mahadevan, A.; Babu, P.P. Activation of Wnt/β-catenin/Tcf signaling pathway in human astrocytomas. Neurochem. Int. 2009, 55, 307–317. [Google Scholar] [CrossRef]
- Utsuki, S.; Sato, Y.; Oka, H.; Tsuchiya, B.; Suzuki, S.; Fujii, K. Relationship between the Expression of E-, N-cadherins and beta-catenin and Tumor Grade in Astrocytomas. J. Neuro-Oncol. 2002, 57, 187–192. [Google Scholar] [CrossRef]
- Zhang, Z.-Q.; Chen, H.-Q.; Chen, Y.-H.; Cheng, X.-F. [Significance of beta-catenin and Cyclin D1 express in glioma]. Chin. J. Cell. Mol. Immunol. 2009, 25, 1010–1012. [Google Scholar]
- Denysenko, T.; Annovazzi, L.; Cassoni, P.; Melcarne, A.; Mellai, M.; Schiffer, D. WNT/β-catenin Signaling Pathway and Downstream Modulators in Low- and High-grade Glioma. Cancer Genom. Proteom. 2015, 13, 31–45. [Google Scholar]
- Liu, X.; Wang, L.; Zhao, S.; Ji, X.; Luo, Y.; Ling, F. β-Catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Med. Oncol. 2010, 28, 608–614. [Google Scholar] [CrossRef]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; et al. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2010, 11, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Fang, J.; Chen, F.; Liu, J.; Wu, J.; Wang, Y.; Song, T.; Zeng, F.; Rao, Y. Downregulation of WIF-1 by Hy-permethylation in Astrocytomas. Acta Biochim. Biophys. Sin. 2010, 42, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Iyer, R.R.; Yu, A.C.H.; Yong, R.L.; Park, D.M.; Weil, R.J.; Ikejiri, B.; Brady, R.O.; Lonser, R.R.; Zhuang, Z. -Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc. Natl. Acad. Sci. USA 2012, 109, 6963–6968. [Google Scholar] [CrossRef] [Green Version]
- Sareddy, G.R.; Pratap, U.P.; Viswanadhapalli, S.; Venkata, P.P.; Nair, B.C.; Krishnan, S.R.; Zheng, S.; Gilbert, A.R.; Brenner, A.J.; Brann, D.W.; et al. PELP1 promotes glioblastoma progression by enhancing Wnt/β-catenin signaling. Neuro-Oncol. Adv. 2019, 1. [Google Scholar] [CrossRef] [Green Version]
- Portela, M.; Casas-Tintó, S. New Cellular Dimensions on Glioblastoma Progression. Neurosci. Insights 2020, 15, 2633105520923076. [Google Scholar] [CrossRef]
- Xu, A.; Yang, H.; Gao, K.; Zhan, Z.; Song, Z.; Huang, T.; Song, Y. Expression profiles and prognostic significance of WNT family members in glioma via bioinformatic analysis. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, X.-Y.; Zhang, X.-Y.; Yuan, F.; Zhao, X.-Y.; You, B.-A. HOXB7 Promotes Proliferation and Metastasis of Glioma by Reg-ulating the Wnt/β-Catenin Pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3146. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ma, Q.; Shu, C.; Xiong, J.; Li, B.; Wu, J.; Zhang, S.; Li, J.; Liu, J.; Wang, J. MicroRNA-301a/ZNRF3/wnt/β-catenin signal regulatory crosstalk mediates glioma progression. Int. J. Oncol. 2020, 58, 45–56. [Google Scholar] [CrossRef]
- Shou, J.; Ali-Osman, F.; Multani, A.S.; Pathak, S.; Fedi, P.; Srivenugopal, K.S. Human Dkk-1, a gene encoding a Wnt antagonist, responds to DNA damage and its overexpression sensitizes brain tumor cells to apoptosis following alkylation damage of DNA. Oncogene 2002, 21, 878–889. [Google Scholar] [CrossRef]
- Roth, W.; Wild-Bode, C.; Platten, M.; Grimmel, C.; Melkonyan, H.S.; Dichgans, J.; Weller, M. Secreted Frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene 2000, 19, 4210–4220. [Google Scholar] [CrossRef] [Green Version]
- Delic, S.; Lottmann, N.; Stelzl, A.; Liesenberg, F.; Wolter, M.; Götze, S.; Zapatka, M.; Shiio, Y.; Sabel, M.C.; Felsberg, J.; et al. MiR-328 promotes glioma cell invasion via SFRP1-dependent Wnt-signaling activation. Neuro-Oncology 2013, 16, 179–190. [Google Scholar] [CrossRef]
- Jin, X.; Jeon, H.-Y.; Joo, K.M.; Kim, J.-K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 Regulates Stemness and Invasiveness of Migrating Glioma Cells Established by Serial Intracranial Transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR Signaling Networks in Glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpel-Massler, G.; Schmidt, U.; Unterberg, A.; Halatsch, M.-E. Therapeutic Inhibition of the Epidermal Growth Factor Receptor in High-Grade Gliomas: Where Do We Stand? Mol. Cancer Res. 2009, 7, 1000–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Chen, L.; Huang, K.; Han, L.; Shi, Z.; Zhang, K.; Pu, P.; Jiang, C.; Kang, C. β-Catenin/Tcf-4 Complex transcriptionally regulates AKT1 in glioma. Int. J. Oncol. 2011, 39, 883–890. [Google Scholar] [CrossRef]
- Kong, D.-S.; Song, S.-Y.; Kim, D.-H.; Joo, K.M.; Yoo, J.-S.; Koh, J.S.; Dong, S.M.; Suh, Y.-L.; Lee, J.-I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2008, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, K.; Shimao, Y.; Sato, S.; Kataoka, H.; Moriyama, T.; Kawano, H.; Wakisaka, S.; Koono, M. Expression of c-Met correlates with grade of malignancy in human astrocytic tumours: An immunohistochemical study. Histopathology 1997, 31, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 97–117. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.; Güttler, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. IDH1R132H mutation causes a less aggressive phenotype and radiosensitizes human malignant glioma cells independent of the oxygenation status. Radiother. Oncol. 2015, 116, 381–387. [Google Scholar] [CrossRef]
- Anson, M.; Crain-Denoyelle, A.-M.; Baud, V.; Chereau, F.; Gougelet, A.; Terris, B.; Yamagoe, S.; Colnot, S.; Viguier, M.; Perret, C.; et al. Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J. Clin. Investig. 2012, 122, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Ren, J.; Shi, J.; Feng, L.; Wang, K.; Zeng, T.; Jin, Y.; Gao, L. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int. J. Biochem. Cell Biol. 2016, 73, 72–81. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumor Biol. 2014, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zuo, H.; Ni, L.; Xia, L.; Zhao, L.; Gong, M.; Nie, D.; Gong, P.; Cui, D.; Shi, W.; et al. An IDH1 mutation inhibits growth of glioma cells via GSH depletion and ROS generation. Neurol. Sci. 2013, 35, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Guillevin, R.; Vallée, J.-N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev. Neurosci. 2017, 29, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Koul, D. PTEN Signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef]
- Birner, P.; Pusch, S.; Christov, C.; Mihaylova, S.; Ms, K.T.; Natchev, S.; Schoppmann, S.F.; Tchorbanov, A.; Streubel, B.; Tuettenberg, J.; et al. Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer 2014, 120, 2440–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; Le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Cai, G.; Yu, Q.; Shen, J.; Gu, Z.; Chen, J.; Shi, W.; Shi, J. IDH1 mutation diminishes aggressive phenotype in glioma stem cells. Int. J. Oncol. 2017, 52, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Mehrjardi, N.Z.; Hänggi, D.; Kahlert, U.D. Current biomarker-associated procedures of cancer modeling-a reference in the context of IDH1 mutant glioma. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1 R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Metellus, P.; Colin, C.; Taieb, D.; Guedj, E.; Nanni-Metellus, I.; De Paula, A.M.; Colavolpe, C.; Fuentes, S.; Dufour, H.; Barrie, M.; et al. IDH mutation status impact on in vivo hypoxia biomarkers expression: New insights from a clinical, nuclear imaging and immunohistochemical study in 33 glioma patients. J. Neuro-Oncol. 2011, 105, 591–600. [Google Scholar] [CrossRef]
- Williams, S.C.; Karajannis, M.A.; Chiriboga, L.; Golfinos, J.G.; Von Deimling, A.; Zagzag, D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1α upregulation in adult glioma. Acta Neuropathol. 2011, 121, 279–281. [Google Scholar] [CrossRef] [Green Version]
- Tchoghandjian, A.; Koh, M.Y.; Taieb, D.; Ganaha, S.; Powis, G.; Bialecki, E.; Graziani, N.; Figarella-Branger, D.; Metellus, P. Hypoxia-associated factor expression in low-grade and anaplastic gliomas: A marker of poor outcome. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Leonard, S.S.; Huang, C.; Vallyathan, V.; Castranova, V.; Shi, X. Role of reactive oxygen species and MAPKs in vanadate-induced G2/M phase arrest. Free Radic. Biol. Med. 2003, 34, 1333–1342. [Google Scholar] [CrossRef]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2006, 26, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Yang, Y.; Han, Y.; Li, X.; Wang, X.; Li, X.; Zhang, Z.; Wang, Y. UCP2 Inhibits ROS-Mediated Apoptosis in A549 under Hypoxic Conditions. PLoS ONE 2012, 7, e30714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, P.; Dey, A.; Sen, R.; Chatterjee, M.; Chattopadhyay, S.; Bandyopadhyay, S.K. Intracellular GSH Depletion Triggered Mitochondrial Bax Translocation to Accomplish Resveratrol-Induced Apoptosis in the U937 Cell Line. J. Pharmacol. Exp. Ther. 2010, 336, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, S.; Maeda, K.; Miki, T.; Mano, J.; Inoue, Y.; Kimura, A. Importance of glucose-6-phosphate dehydrogenase in the adaptive response to hydrogen peroxide in Saccharomyces cerevisiae. Biochem. J. 1998, 330, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Gatenby, R.A. Metabolism and Its Sequelae in Cancer Evolution and Therapy. Cancer J. 2015, 21, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Gatenby, R.A. Adaptive landscapes and emergent phenotypes: Why do cancers have high glycolysis? J. Bioenerg. Biomembr. 2007, 39, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer. 2018, 1870, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Luo, J.; Rana, J.S.; Laham, R.; Sellke, F.W.; Li, J. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc. Res. 2006, 69, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airley, R.E.; Mobasheri, A. Hypoxic Regulation of Glucose Transport, Anaerobic Metabolism and Angiogenesis in Cancer: Novel Pathways and Targets for Anticancer Therapeutics. Chemotherapy 2007, 53, 233–256. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Guo, S.; Yang, L. All-trans retinoic acid upregulates VEGF expression in glioma cells in vitro. J. Biomed. Res. 2013, 27, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Suk, K. Pyruvate Dehydrogenase Kinase as a Potential Therapeutic Target for Malignant Gliomas. Brain Tumor Res. Treat. 2013, 1, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Maxwell, P. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubuk, C.; Hidalgo, M.R.; Amadoz, A.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Gene Expression Integration into Pathway Modules Reveals a Pan-Cancer Metabolic Landscape. Cancer Res. 2018, 78, 6059–6072. [Google Scholar] [CrossRef] [Green Version]
- Çubuk, C.; Hidalgo, M.R.; Amadoz, A.; Rian, K.; Salavert, F.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Differential metabolic activity and discovery of therapeutic targets using summarized metabolic pathway models. NPJ Syst. Biol. Appl. 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr. Metab. 2005, 2, 30. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineura, K.; Yasuda, T.; Kowada, M.; Shishido, F.; Ogawa, T.; Uemura, K. Positron emission tomographic evaluation of histological malignancy in gliomas using oxygen-15 and fluorine-18-fluorodeoxyglucose. Neurol. Res. 1986, 8, 164–168. [Google Scholar] [CrossRef]
- Oudard, S.; Arvelo, F.; Miccoli, L.; Apiou, F.; Dutrillaux, A.M.; Poisson, M.; Dutrillaux, B.; Poupon, M.F. High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss. Br. J. Cancer 1996, 74, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Constant, J.S.; Feng, J.J.; Zabel, D.D.; Yuan, H.; Suh, D.Y.; Scheuenstuhl, H.; Hunt, T.K.; Hussain, M.Z. Lactate elicits vascular endothelial growth factor from macrophages: A possible alternative to hypoxia. Wound Repair Regen. 2000, 8, 353–360. [Google Scholar] [CrossRef]
- Thompson, C.B. Wnt meets Warburg: Another piece in the puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1 plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and Metastasis in Colorectal Cancer: Epithelial-Mesenchymal Transition, Mesenchymal-Epithelial Transition, Stem Cells and β-Catenin. Cells Tissues Organs 2005, 179, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.; Garner, C.; Digman, A.M.; Teitell, A.M.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Chafey, P.; Finzi, L.; Boisgard, R.; Caüzac, M.; Clary, G.; Broussard, C.; Pégorier, J.-P.; Guillonneau, F.; Mayeux, P.; Camoin, L.; et al. Proteomic analysis of β-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 2009, 9, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Rethinking the Warburg Effect with Myc Micromanaging Glutamine Metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Moon, R.T. The Promise and Perils of Wnt Signaling Through Beta-Catenin. Science 2002, 296, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Acebron, S.P. Mitotic and mitogenic Wnt signalling. EMBO J. 2012, 31, 2705–2713. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The Beta-Catenin/TCF-4 Complex Imposes a Crypt Progenitor Phenotype on Colorectal Cancer Cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Popescu, A.M.; Purcaru, S.O.; Alexandru, O.; Dricu, A. New perspectives in glioblastoma antiangiogenic therapy. Współczesna Onkol. 2016, 20, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Da Lee, R.; Kang, S.-K.; Han, S.Y.; Park, K.L.; Yang, K.H.; Song, Y.S.; Park, H.J.; Lee, Y.M.; Yun, Y.P.; et al. Neuronal differentiation of embryonic midbrain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp. Cell Res. 2004, 297, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.-H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P.S. Epidermal Growth Factor Receptor: A Novel Target of the Wnt/Beta-Catenin Pathway in Liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Lan, F.; Yang, W.; Yang, Y.; Han, L.; Zhang, A.; Liu, J.; Zeng, H.; Jiang, T.; Pu, P.; et al. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010, 1366, 27–37. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, K.; Shi, Z.; Zou, J.; Wang, Y.; Jia, Z.; Zhang, A.; Han, L.; Yue, X.; Liu, N.; et al. High -catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro-Oncology 2011, 13, 600–609. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically Derived Lactate Stimulates Revascularization and Tissue Repair via Redox Mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.E.; Lee, H.; Cho, I.; Chung, D.H.; Yoon, S.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.; Ye, S.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, C.; Zhang, W.; Zhang, G.; Zhao, X.; Yang, S.; Wang, Y.; Lu, N.; Zhu, H.; Xu, N. β-Catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008, 271, 85–97. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekanty, A.; Lavista-Llanos, S.; Irisarri, M.; Oldham, S.; Wappner, P. The insulin-PI3K/TOR pathway induces a HIF-dependent transcriptional response in Drosophila by promoting nuclear localization of HIF-α/Sima. J. Cell Sci. 2005, 118, 5431–5441. [Google Scholar] [CrossRef] [Green Version]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Lactate Stimulates Vasculogenic Stem Cells via the Thioredoxin System and Engages an Autocrine Activation Loop Involving Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 2008, 28, 6248–6261. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G. TSC2 Regulates VEGF through MTOR-Dependent and -Independent Pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Land, S.C.; Tee, A.R. Hypoxia-inducible Factor 1α Is Regulated by the Mammalian Target of Rapamycin (mTOR) via an mTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [Green Version]
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential Dependence of Hypoxia-inducible Factors 1α and 2α on mTORC1 and mTORC2. J. Biol. Chem. 2008, 283, 34495–34499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for Direct Activation of mTORC2 Kinase Activity by Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef] [Green Version]
- Kaur, B.; Tan, C.; Brat, D.J.; Van Meir, E.G. Genetic and hypoxic regulation of angiogenesis in gliomas. J. Neuro-Oncology 2004, 70, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parliament, M.B.; Allalunis-Turner, M.J.; Franko, A.J.; Olive, P.L.; Mandyam, R.; Santos, C.; Wolokoff, B. Vascular endothelial growth factor expression is independent of hypoxia in human malignant glioma spheroids and tumours. Br. J. Cancer 2000, 82, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2009, 29, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Zhong, H.; Scalzitti, J.M.; Laughner, E.; Simons, J.W.; Semenza, G.L. Expression of hypoxia-inducible factor 1alpha in brain tumors: Association with angiogenesis, invasion, and progression. Cancer 2000, 88, 2606–2618. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Unruh, A.; Ressel, A.; Mohamed, H.G.; Johnson, R.S.; Nadrowitz, R.; Richter, E.; Katschinski, D.M.; Wenger, R.H. The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene 2003, 22, 3213–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2015, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohil, K.; Brooks, G.A. Exercise tames the wild side of the Myc network: A hypothesis. Am. J. Physiol. Metab. 2012, 303, E18–E30. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Turley, H.; Harris, A.L.; Koukourakis, M.I. Lactate dehydrogenase 5 (LDH-5) expression in endometrial cancer relates to the activated VEGF/VEGFR2(KDR) pathway and prognosis. Gynecol. Oncol. 2006, 103, 912–918. [Google Scholar] [CrossRef]
- Kolev, Y.; Uetake, H.; Takagi, Y.; Sugihara, K. Lactate Dehydrogenase-5 (LDH-5) Expression in Human Gastric Cancer: Association with Hypoxia-Inducible Factor (HIF-1α) Pathway, Angiogenic Factors Production and Poor Prognosis. Ann. Surg. Oncol. 2008, 15, 2336–2344. [Google Scholar] [CrossRef]
- Koukourakis, I.M.; Giatromanolaki, A.; Sivridis, E.; Bougioukas, G.; Didilis, V.; Gatter, K.C.; Harris, A.L.; for the ‘Tumour and Angiogenesis Research Group’. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br. J. Cancer 2003, 89, 877–885. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and Predictive Role of Lactate Dehydrogenase 5 Expression in Colorectal Cancer Patients Treated with PTK787/ZK 222584 (Vatalanib) Antiangiogenic Therapy. Clin. Cancer Res. 2011, 17, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Dang, C.V. Cancer’s Molecular Sweet Tooth and the Warburg Effect: Figure 1. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic Regulation of Lactate Dehydrogenase A. Interaction between Hypoxia-Inducible Factor 1 and CAMP Response Elements. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.C.; Shim, H.; Li, Q.; Wu, C.S.; Lee, A.L.; Maity, A.; Dang, C.V. Identification of putative c-Myc-responsive genes: Characterization of RCL, a novel growth-related gene. Mol. Cell. Biol. 1997, 17, 4967–4978. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Jiang, M.; Chen, Z.; Xu, X.; Hu, H.; Zhao, X.; Gao, X.; Guo, L. Lactate Dehydrogenase 5 Expression in Non-Hodgkin Lymphoma Is Associated with the Induced Hypoxia Regulated Protein and Poor Prognosis. PLoS ONE 2013, 8, e74853. [Google Scholar] [CrossRef] [PubMed]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2016, 38, 119–133. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible Inactivation of HIF-1 Prolyl Hydroxylases Allows Cell Metabolism to Control Basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Königsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate stimulates endothelial cell migration. Wound Repair Regen. 2006, 14, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer: Figure 1. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic Extracellular pH Induces Vascular Endothelial Growth Factor (VEGF) in Human Glioblastoma Cells via ERK1/2 MAPK Signaling Pathway. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Fidler, I.J. Acidic PH-Induced Elevation in Interleukin 8 Expression by Human Ovarian Carcinoma Cells. Cancer Res. 2000, 60, 4610–4616. [Google Scholar] [PubMed]
- Haaga, J.R.; Haaga, R. Acidic lactate sequentially induced lymphogenesis, phlebogenesis, and arteriogenesis (ALPHA) hypothesis: Lactate-triggered glycolytic vasculogenesis that occurs in normoxia or hypoxia and complements the traditional concept of hypoxia-based vasculogenesis. Surgery 2013, 154, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Trabold, O.; Wagner, S.; Wicke, C.; Bs, H.S.; Hussain, M.Z.; Rosen, N.; Bs, A.S.; Becker, H.D.; Hunt, T.K. Lactate and oxygen constitute a fundamental regulatory mechanism in wound healing. Wound Repair Regen. 2003, 11, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Marker | Cell Pathways | Regulation | Subtypes | Prognosis | References |
|---|---|---|---|---|---|
| IDH1 mutations | NADPH-dependent reduction of alpha-ketoglutarate | HIF-1α reduction activity | Glioblastoma | Good survival rate | [136] |
| IDH1 mutations | TP 53 and total 1p/19q deletions | - | Oligodendroglia tumors | Good survival rate | [26] |
| IDH1-R132H mutation | β-catenin, TCF4 and LEF1 downregulation | Decrease WNT signaling | Glioma cells | Lower invasion | [126,134] |
| IDH1-R132H mutation | PI3K/Akt pathway decrease | Decrease WNT signaling | Glioma cells | Good survival rate | [131,132,133] |
| IDH1 mutations | HIF-1α reduction activity | Decrease WNT signaling | Restricted to necrotic areas | Poor survival rate | [137,138,139] |
| EGFR | β-catenin accumulation | Increase WNT signaling | Astrocytomas | Glioma progression and invasion | [98,99,101,102,103,104,105] |
| sFRP1 depletion | β-catenin accumulation | Increase WNT signaling | Glioblastoma | Inhibit Motility and Promote Growth | [112,113] |
| RTKs | WNT signaling | IDH1 decrease | Glioblastoma | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Opposed Interplay between IDH1 Mutations and the WNT/β-Catenin Pathway: Added Information for Glioma Classification. Biomedicines 2021, 9, 619. https://doi.org/10.3390/biomedicines9060619
Vallée A, Lecarpentier Y, Vallée J-N. Opposed Interplay between IDH1 Mutations and the WNT/β-Catenin Pathway: Added Information for Glioma Classification. Biomedicines. 2021; 9(6):619. https://doi.org/10.3390/biomedicines9060619
Chicago/Turabian StyleVallée, Alexandre, Yves Lecarpentier, and Jean-Noël Vallée. 2021. "Opposed Interplay between IDH1 Mutations and the WNT/β-Catenin Pathway: Added Information for Glioma Classification" Biomedicines 9, no. 6: 619. https://doi.org/10.3390/biomedicines9060619