
Neuroinflammation in Ischemic Stroke: Inhibition of cAMP-Specific Phosphodiesterases (PDEs) to the Rescue


 and
and
Abstract
:1. Introduction
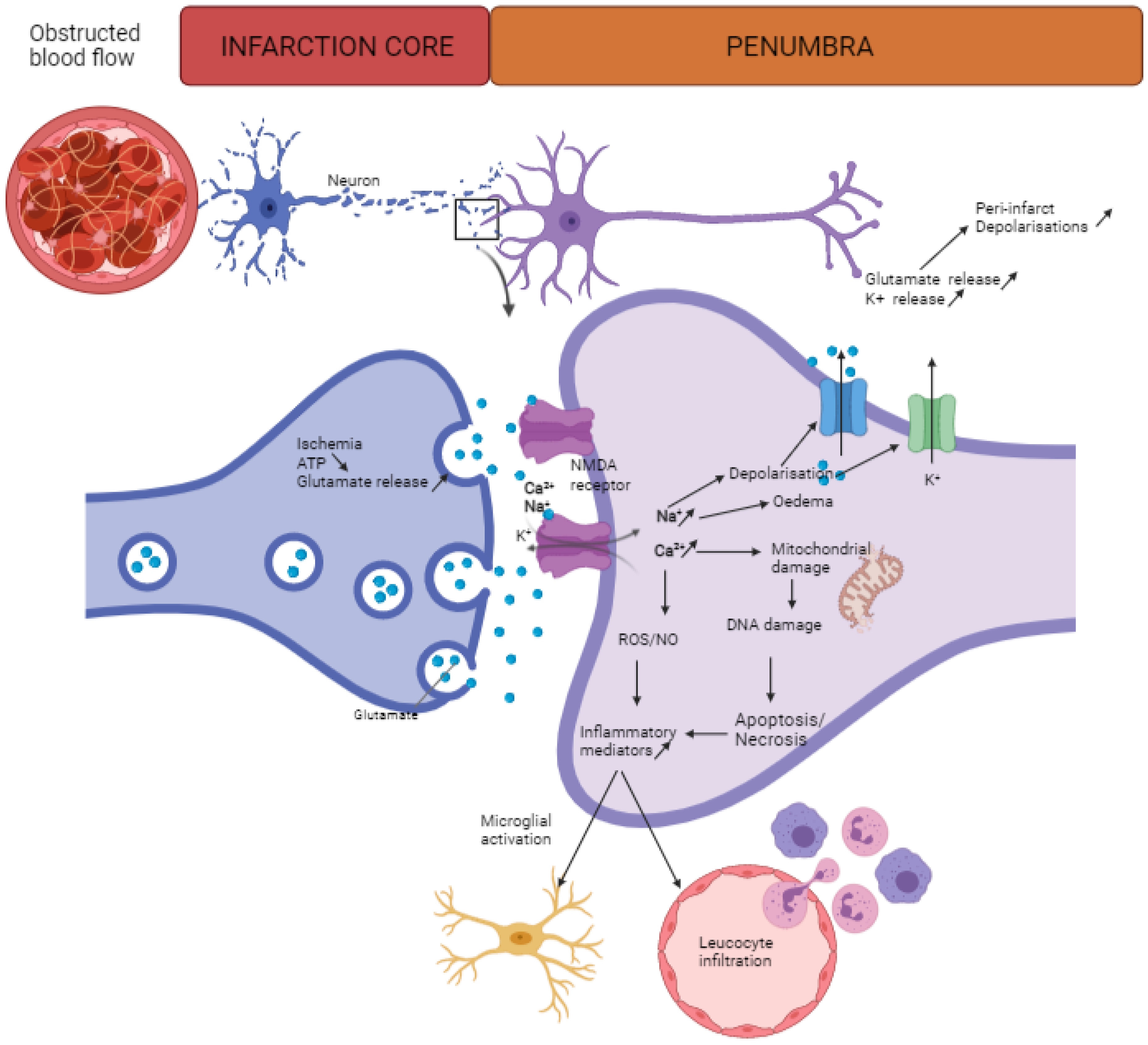
2. Neuroinflammation after Stroke
2.1. Innate Immune Cells
2.2. Adaptive Immune Cells
3. cAMP
4. cAMP-Specific PDE Inhibition as Therapy for Neuroinflammation in Ischemic Stroke
4.1. PDE4 Inhibition
4.2. PDE7 Inhibition
4.3. PDE8 Inhibition
4.4. Functional Read-Outs
4.5. Reflection on the Potential of PDE Inhibition in Other Neuroinflammatory-Related Disorders, Such as Traumatic Brain Injury, Alzheimer’s Disease, and Multiple Sclerosis
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lindsay, M.P.; Norrving, B.; Sacco, R.L.; Brainin, M.; Hacke, W.; Martins, S.; Pandian, J.; Feigin, V. World Stroke Organization (WSO): Global stroke fact sheet 2019. Int. J. Stroke 2019, 14, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S. Stroke in the 21(st) century: A snapshot of the burden, epidemiology, and quality of life. Stroke Res. Treat 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankey, G.J.; Jamrozik, K.; Broadhurst, R.J.; Forbes, S.; Anderson, C.S. Long-term disability after first-ever stroke and related prognostic factors in the Perth Community Stroke Study, 1989–1990. Stroke 2002, 33, 1034–1040. [Google Scholar] [CrossRef] [Green Version]
- Jauch, E.C.; Cucchiara, B.; Adeoye, O.; Meurer, W.; Brice, J.; Chan, Y.Y.; Gentile, N.; Hazinski, M.F. Part 11: Adult stroke: 2010 american heart association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2010, 122, S818–S828. [Google Scholar] [CrossRef] [Green Version]
- Herpich, F.; Rincon, F. Management of acute ischemic stroke. Crit. Care Med. 2020, 48, 1654–1663. [Google Scholar] [CrossRef]
- Jones, A.; O’Connell, N.; David, A.S.; Chalder, T. Functional stroke symptoms: A narrative review and conceptual model. J. Neuropsychiatry Clin. Neurosci. 2020, 32, 14–23. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers. 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, R.R.; Baron, J.C. Pathophysiology of ischaemic stroke: Insights from imaging, and implications for therapy and drug discovery. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S44–S54. [Google Scholar] [CrossRef] [Green Version]
- Musuka, T.D.; Wilton, S.B.; Traboulsi, M.; Hill, M.D. Diagnosis and management of acute ischemic stroke: Speed is critical. CMAJ 2015, 187, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Hui, C.; Tadi, P.; Patti, L. Ischemic Stroke; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Khandelwal, P.; Yavagal, D.R.; Sacco, R.L. Acute ischemic stroke intervention. J. Am. Coll. Cardiol. 2016, 67, 2631–2644. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and stroke: An overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Paciaroni, M.; Caso, V.; Agnelli, G. The concept of ischemic penumbra in acute stroke and therapeutic opportunities. Eur. Neurol. 2009, 61, 321–330. [Google Scholar] [CrossRef]
- Kaufmann, A.M.; Firlik, A.D.; Fukui, M.B.; Wechsler, L.R.; Jungries, C.A.; Yonas, H. Ischemic core and penumbra in human stroke. Stroke 1999, 30, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanese, L.; Tarsia, J.; Fisher, M. Acute ischemic stroke therapy overview. Circ. Res. 2017, 120, 541–558. [Google Scholar] [CrossRef]
- Liaw, N.; Liebeskind, D. Emerging therapies in acute ischemic stroke. F1000Res 2020, 9. [Google Scholar] [CrossRef]
- Neuhaus, A.A.; Couch, Y.; Hadley, G.; Buchan, A.M. Neuroprotection in stroke: The importance of collaboration and reproducibility. Brain 2017, 140, 2079–2092. [Google Scholar] [CrossRef] [Green Version]
- Cocho, D.; Belvís, R.; Martí-Fàbregas, J.; Molina-Porcel, L.; Díaz-Manera, J.; Aleu, A.; Pagonabarraga, J.; García-Bargo, D.; Mauri, A.; Martí-Vilalta, J.L. Reasons for exclusion from thrombolytic therapy following acute ischemic stroke. Neurology 2005, 64, 719–720. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaerschalk, B.M.; Kleindorfer, D.O.; Adeoye, O.M.; Demchuk, A.M.; Fugate, J.E.; Grotta, J.C.; Khalessi, A.A.; Levy, E.I.; Palesch, Y.Y.; Prabhakaran, S.; et al. Scientific rationale for the inclusion and exclusion criteria for intravenous alteplase in acute ischemic stroke: A statement for healthcare professionals from the American heart association/American stroke association. Stroke 2016, 47, 581–641. [Google Scholar] [CrossRef] [PubMed]
- Fugate, J.E.; Rabinstein, A.A. Absolute and relative contraindications to IV rt-PA for acute ischemic stroke. Neurohospitalist 2015, 5, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lioutas, V.A.; Novak, V. Intranasal insulin neuroprotection in ischemic stroke. Neural Regen Res. 2016, 11, 400–401. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Devel. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Sarmah, D.; Kalia, K.; Borah, A.; Dave, K.R.; Yavagal, D.R.; Bhattacharya, P. Animal Models of Ischemic Stroke. In Application of Biomedical Engineering in Neuroscience; Paul, S., Ed.; Springer: Singapore, 2019; pp. 41–45. [Google Scholar] [CrossRef]
- McBride, D.W.; Zhang, J.H. Precision stroke animal models: The permanent MCAO model should be the primary model, Not transient MCAO. Transl. Stroke Res. 2017. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Nilupul Perera, M.; Ma, H.K.; Arakawa, S.; Howells, D.W.; Markus, R.; Rowe, C.C.; Donnan, G.A. Inflammation following stroke. J. Clin. Neurosci. 2006, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front Immunol. 2019, 10, 2167. [Google Scholar] [CrossRef] [Green Version]
- Planas, A.M. Role of immune cells migrating to the ischemic brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. The inflammatory response after ischemic stroke: Targeting β(2) and β(1) integrins. Front. Neurosci. 2019, 13, 540. [Google Scholar] [CrossRef]
- Kim, E.; Cho, S. Microglia and Monocyte-Derived Macrophages in Stroke. Neurotherapeutics. 2016, 13, 702–718. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Lei, T.Y.; Ye, Y.Z.; Zhu, X.Q.; Smerin, D.; Gu, L.J.; Xiong, X.X.; Zhang, H.F.; Jian, Z.H. The immune response of T cells and therapeutic targets related to regulating the levels of T helper cells after ischaemic stroke. J. Neuroinflamm. 2021, 18, 25. [Google Scholar] [CrossRef]
- Na, S.Y.; Mracsko, E.; Liesz, A.; Hünig, T.; Veltkamp, R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke 2015, 46, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Zhou, W.; Na, S.Y.; Hämmerling, G.J.; Garbi, N.; Karcher, S.; Mracsko, E.; Backs, J.; Rivest, S.; Veltkamp, R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J. Neurosci. 2013, 33, 17350–17362. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 2009, 158, 70–86. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Feng, J.; Hao, Y.; You, J.; Wang, X.; Yin, X.; Shang, P.; Ma, D. Cyclic adenosine monophosphate in acute ischemic stroke: Some to update, more to explore. J. Neurol. Sci. 2020, 413, 116775. [Google Scholar] [CrossRef]
- Saponaro, A.; Cantini, F.; Porro, A.; Bucchi, A.; DiFrancesco, D.; Maione, V.; Donadoni, C.; Introini, B.; Mesirca, P.; Mangoni, M.E.; et al. A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. eLife 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Saand, M.A.; Xu, Y.P.; Li, W.; Wang, J.P.; Cai, X.Z. Cyclic nucleotide gated channel gene family in tomato: Genome-wide identification and functional analyses in disease resistance. Front. Plant Sci. 2015, 6, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.D.; Kessler, K.M.; Pincheira, R.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor activates CRE-binding protein by signaling through the KDR receptor tyrosine kinase. J. Biol. Chem. 2001, 276, 25184–25189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007, 274, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Rao, G.N. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arter. Thromb. Vasc. Biol. 2009, 29, 809–815. [Google Scholar] [CrossRef]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Murray, A.J.; Shewan, D.A. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol. Cell Neurosci. 2008, 38, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Peace, A.G.; Shewan, D.A. New perspectives in cyclic AMP-mediated axon growth and guidance: The emerging epoch of Epac. Brain Res. Bull 2011, 84, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, K.; Mehta, S.; Ramamurthy, S.; Ronnett, G.V.; Zhou, F.Q.; Zhang, J. AKAP-mediated feedback control of cAMP gradients in developing hippocampal neurons. Nat. Chem. Biol. 2017, 13, 425–431. [Google Scholar] [CrossRef]
- Wang, A.R.; Hu, M.Z.; Zhang, Z.L.; Zhao, Z.Y.; Li, Y.B.; Liu, B. Fastigial nucleus electrostimulation promotes axonal regeneration after experimental stroke via cAMP/PKA pathway. Neurosci. Lett. 2019, 699, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kitagawa, K.; Omura-Matsuoka, E.; Todo, K.; Terasaki, Y.; Sugiura, S.; Hatazawa, J.; Yagita, Y.; Hori, M. The phosphodiesterase inhibitor rolipram promotes survival of newborn hippocampal neurons after ischemia. Stroke 2007, 38, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.; Soares, L.M.; Schepers, M.; Milani, H.; Vanmierlo, T.; Prickaerts, J.; Weffort de Oliveira, R.M. Roflumilast promotes memory recovery and attenuates white matter injury in aged rats subjected to chronic cerebral hypoperfusion. Neuropharmacology 2018, 138, 360–370. [Google Scholar] [CrossRef]
- Schubert, P.; Morino, T.; Miyazaki, H.; Ogata, T.; Nakamura, Y.; Marchini, C.; Ferroni, S. Cascading glia reactions: A common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann. N. Y. Acad. Sci. 2000, 903, 24–33. [Google Scholar] [CrossRef]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.Y.; Zhang, S.; Gao, Y.; Wang, Z.Z.; Chen, N.H. Selective modulation of microglia polarization to M2 phenotype for stroke treatment. Int. Immunopharmacol. 2015, 25, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, S.; Marchini, C.; Schubert, P.; Rapisarda, C. Two distinct inwardly rectifying conductances are expressed in long term dibutyryl-cyclic-AMP treated rat cultured cortical astrocytes. FEBS Lett. 1995, 367, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Sasaki, K.; Ifuku, M.; Wada, K. Multifunctional effects of bradykinin on glial cells in relation to potential anti-inflammatory effects. Neurochem. Int. 2007, 51, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, S.; Wang, S.J.; Yu, C.; Paganini-Hill, A.; Fisher, M.J. Tissue plasminogen activator expression and barrier properties of human brain microvascular endothelial cells. Cell Physiol. Biochem. 2011, 28, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Harris, G.J.; Pinder, E.M.; Macfarlane, J.G.; Hellyer, T.P.; Rostron, A.J.; Conway Morris, A.; Thickett, D.R.; Perkins, G.D.; McAuley, D.F.; et al. Exchange protein directly activated by cyclic AMP (EPAC) activation reverses neutrophil dysfunction induced by β2-agonists, corticosteroids, and critical illness. J. Allergy Clin. Immunol. 2016, 137, 535–544. [Google Scholar] [CrossRef]
- Ernens, I.; Rouy, D.; Velot, E.; Devaux, Y.; Wagner, D.R. Adenosine inhibits matrix metalloproteinase-9 secretion by neutrophils: Implication of A2a receptor and cAMP/PKA/Ca2+ pathway. Circ. Res. 2006, 99, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Jackson, E.K.; Ren, J.; Mi, Z. Extracellular 2′,3′-cAMP is a source of adenosine. J. Biol. Chem. 2009, 284, 33097–33106. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kotova, E.; Chen, Z.S.; Lee, K.; Hopper-Borge, E.; Belinsky, M.G.; Kruh, G.D. MRP8, ATP-binding cassette C11 (ABCC11), is a cyclic nucleotide efflux pump and a resistance factor for fluoropyrimidines 2′,3′-dideoxycytidine and 9′-(2′-phosphonylmethoxyethyl)adenine. J. Biol. Chem. 2003, 278, 29509–29514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar] [CrossRef] [Green Version]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russel, F.G.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Pugliese, A.M.; Pedata, F. Adenosine receptors in cerebral ischemia. Int. Rev. Neurobiol. 2014, 119, 309–348. [Google Scholar] [CrossRef] [PubMed]
- Blokland, A.; Heckman, P.; Vanmierlo, T.; Schreiber, R.; Paes, D.; Prickaerts, J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol. Sci. 2019, 40, 971–985. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Paes, D.S.M.; Rombaut, B.; van den Hove, D.; Vanmierlo, T.; Prickaerts, J. The molecular biology of PDE4 enzymes as pharmacological targets: An interplay of isoforms, conformational states, and inhibitors. Pharmacol. Rev. 2021, in press. [Google Scholar]
- Kraft, P.; Schwarz, T.; Göb, E.; Heydenreich, N.; Brede, M.; Meuth, S.G.; Kleinschnitz, C. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp. Neurol. 2013, 247, 80–90. [Google Scholar] [CrossRef]
- Yang, F.; Sumbria, R.K.; Xue, D.; Yu, C.; He, D.; Liu, S.; Paganini-Hill, A.; Fisher, M. Effects of PDE4 pathway inhibition in rat experimental stroke. J. Pharm. Pharm. Sci. 2014, 17, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Xu, J.; Cai, N.; Li, M.; Liu, L.; Qin, Y.; Li, X.; Wang, H. Roflumilast prevents ischemic stroke-induced neuronal damage by restricting GSK3β-mediated oxidative stress and IRE1α/TRAF2/JNK pathway. Free Radic. Biol. Med. 2021, 163, 281–296. [Google Scholar] [CrossRef]
- Xu, B.; Wang, T.; Xiao, J.; Dong, W.; Wen, H.Z.; Wang, X.; Qin, Y.; Cai, N.; Zhou, Z.; Xu, J.; et al. FCPR03, a novel phosphodiesterase 4 inhibitor, alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β/ β-catenin signaling pathway. Biochem. Pharmacol. 2019, 163, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, H.; Zhong, J.; Feng, H.; Wang, H.; Cheng, Y.; Zou, Z.; Huang, C.; Zhou, Z.; Zheng, W.; et al. The phosphodiesterase-4 inhibitor, FCPR16, attenuates ischemia-reperfusion injury in rats subjected to middle cerebral artery occlusion and reperfusion. Brain Res. Bull. 2018, 137, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, E.R.; Schepers, M.; Keiko, J.; Kunieda, C.; Milhani, H.; Vanmierlo, T.; Prickaerts, J.; Weffort de Oliveira, R.M. Positive effects of roflumilast on behavior, neuroinflammation, and white matter injury in mice with global cerebral ischemia. Behav. Pharmacol. 2021, in press. [Google Scholar]
- Miller, A.D.; Leslie, R.A. The area postrema and vomiting. Front Neuroendocr. 1994, 15, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Bonham, A.C.; Hasser, E.M. Area postrema and aortic or vagal afferents converge to excite cells in nucleus tractus solitarius. Am. J. Physiol. 1993, 264, H1674–H1685. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.O.; Briggs, D.B.; Knox, A.P.; Strominger, N. Excitation of area postrema neurons by transmitters, peptides, and cyclic nucleotides. J. Neurophysiol. 1988, 59, 358–369. [Google Scholar] [CrossRef]
- Cherry, J.A.; Davis, R.L. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J. Comp. Neurol. 1999, 407, 287–301. [Google Scholar] [CrossRef]
- Takahashi, M.; Terwilliger, R.; Lane, C.; Mezes, P.S.; Conti, M.; Duman, R.S. Chronic antidepressant administration increases the expression of cAMP-specific phosphodiesterase 4A and 4B isoforms. J. Neurosci. 1999, 19, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Mori, F.; Pérez-Torres, S.; De Caro, R.; Porzionato, A.; Macchi, V.; Beleta, J.; Gavaldà, A.; Palacios, J.M.; Mengod, G. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J. Chem. Neuroanat. 2010, 40, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, A.; Savoie, C.; Stamatiou, P.B.; Lachance, N.; Jolicoeur, P.; Rasori, R.; Chan, C.C. Assessing the emetic potential of PDE4 inhibitors in rats. Br. J. Pharmacol. 2002, 135, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Vanmierlo, T.; Creemers, P.; Akkerman, S.; van Duinen, M.; Sambeth, A.; De Vry, J.; Uz, T.; Blokland, A.; Prickaerts, J. The PDE4 inhibitor roflumilast improves memory in rodents at non-emetic doses. Behav. Brain Res. 2016, 303, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Nelissen, E.; van Goethem, N.P.; Bonassoli, V.T.; Heckman, P.R.A.; van Hagen, B.T.J.; Suay, D.; Wouters, C.; Prickaerts, J. Validation of the xylazine/ketamine anesthesia test as a predictor of the emetic potential of pharmacological compounds in rats. Neurosci. Lett. 2019, 699, 41–46. [Google Scholar] [CrossRef]
- Robichaud, A.; Savoie, C.; Stamatiou, P.B.; Tattersall, F.D.; Chan, C.C. PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology 2001, 40, 262–269. [Google Scholar] [CrossRef]
- Robichaud, A.; Stamatiou, P.B.; Jin, S.L.; Lachance, N.; MacDonald, D.; Laliberté, F.; Liu, S.; Huang, Z.; Conti, M.; Chan, C.C. Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J. Clin. Investig. 2002, 110, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Z.; Cheng, Y.F.; Zou, Z.Q.; Ge, B.C.; Yu, H.; Huang, C.; Wang, H.T.; Yang, X.M.; Xu, J.P. Discovery of N-Alkyl Catecholamides as Selective Phosphodiesterase-4 Inhibitors with Anti-neuroinflammation Potential Exhibiting Antidepressant-like Effects at Non-emetic Doses. ACS Chem. Neurosci. 2017, 8, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Bruno, O.; Fedele, E.; Prickaerts, J.; Parker, L.A.; Canepa, E.; Brullo, C.; Cavallero, A.; Gardella, E.; Balbi, A.; Domenicotti, C.; et al. GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br. J. Pharmacol. 2011, 164, 2054–2063. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Brullo, C.; Prickaerts, J.; Arancio, O.; Villa, C.; Rebosio, C.; Calcagno, E.; Balbi, M.; van Hagen, B.T.; Argyrousi, E.K.; et al. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer’s disease. Sci. Rep. 2017, 7, 46320. [Google Scholar] [CrossRef] [PubMed]
- Campos-Toimil, M.; Keravis, T.; Orallo, F.; Takeda, K.; Lugnier, C. Short-term or long-term treatments with a phosphodiesterase-4 (PDE4) inhibitor result in opposing agonist-induced Ca2+ responses in endothelial cells. Br. J. Pharmacol. 2008, 154, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Bonato, J.M.; Meyer, E.; de Mendonça, P.S.B.; Milani, H.; Prickaerts, J.; Weffort de Oliveira, R.M. Roflumilast protects against spatial memory impairments and exerts anti-inflammatory effects after transient global cerebral ischemia. Eur. J. Neurosci. 2021, 53, 1171–1188. [Google Scholar] [CrossRef]
- Soares, L.M.; De Vry, J.; Steinbusch, H.W.M.; Milani, H.; Prickaerts, J.; Weffort de Oliveira, R.M. Rolipram improves cognition, reduces anxiety- and despair-like behaviors and impacts hippocampal neuroplasticity after transient global cerebral ischemia. Neuroscience 2016, 326, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Tibbo, A.J.; Baillie, G.S. Phosphodiesterase 4B: Master Regulator of Brain Signaling. Cells 2020, 9, 1254. [Google Scholar] [CrossRef]
- Brain RNA-Seq. Available online: https://brainrnaseq.org/ (accessed on 1 May 2021).
- Yue, X.; Lixia, L.; Yan, H.; Zhang, P.; Gui, Y.; Song, J. Association between PDE4D polymorphism and ischemic stroke in young population. Saudi. J. Biol. Sci. 2019, 26, 1023–1026. [Google Scholar] [CrossRef]
- Xue, H.; Wang, H.; Song, X.; Li, W.; Sun, K.; Zhang, W.; Wang, X.; Wang, Y.; Hui, R. Phosphodiesterase 4D gene polymorphism is associated with ischaemic and haemorrhagic stroke. Clin. Sci. 2009, 116, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ding, R.; Kuang, P.; Wang, L.; Deng, H.; Xiong, Q.; Jiang, H. Interaction between CONNEXIN37 and PDE4D gene polymorphisms with susceptibility to ischemic stroke in Chinese population. Exp. Biol. Med. 2019, 244, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Zhang, D.; Mang, J.; He, J.; Liu, H.; Shao, Y.; Han, F.; Xu, Z. Association between phosphodiesterase 4D (PDE4D) SNP 87 and ischemic stroke: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 1715–1725. [Google Scholar] [PubMed]
- Domingues-Montanari, S.; Fernández-Cadenas, I.; del Rio-Espinola, A.; Corbeto, N.; Krug, T.; Manso, H.; Gouveia, L.; Sobral, J.; Mendioroz, M.; Fernández-Morales, J.; et al. Association of a genetic variant in the ALOX5AP with higher risk of ischemic stroke: A case-control, meta-analysis and functional study. Cereb. Dis. 2010, 29, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Bevan, S.; Dichgans, M.; Gschwendtner, A.; Kuhlenbäumer, G.; Ringelstein, E.B.; Markus, H.S. Variation in the PDE4D gene and ischemic stroke risk: A systematic review and meta-analysis on 5200 cases and 6600 controls. Stroke 2008, 39, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gretarsdottir, S.; Thorleifsson, G.; Reynisdottir, S.T.; Manolescu, A.; Jonsdottir, S.; Jonsdottir, T.; Gudmundsdottir, T.; Bjarnadottir, S.M.; Einarsson, O.B.; Gudjonsdottir, H.M.; et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat. Genet. 2003, 35, 131–138. [Google Scholar] [CrossRef]
- Smith, S.J.; Brookes-Fazakerley, S.; Donnelly, L.E.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2003, 284, L279–L289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Cieslinski, L.B.; Newton, R.; Donnelly, L.E.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: In vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol. Pharmacol. 2004, 66, 1679–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yee, C.; Beavo, J.A. CD3- and CD28-dependent induction of PDE7 required for T cell activation. Science 1999, 283, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wyman, A.R.; Alaamery, M.A.; Argueta, S.A.; Ivey, F.D.; Meyers, J.A.; Lerner, A.; Burdo, T.H.; Connolly, T.; Hoffman, C.S.; et al. Anti-inflammatory effects of novel barbituric acid derivatives in T lymphocytes. Int. Immunopharmacol. 2016, 38, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kadoshima-Yamaoka, K.; Murakawa, M.; Goto, M.; Tanaka, Y.; Inoue, H.; Murafuji, H.; Nagahira, A.; Hayashi, Y.; Nagahira, K.; Miura, K.; et al. ASB16165, a novel inhibitor for phosphodiesterase 7A (PDE7A), suppresses IL-12-induced IFN-gamma production by mouse activated T lymphocytes. Immunol. Lett. 2009, 122, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; McIntyre, K.W.; Townsend, R.M.; Shen, H.H.; Pitts, W.J.; Dodd, J.H.; Nadler, S.G.; McKinnon, M.; Watson, A.J. Phosphodiesterase 7A-deficient mice have functional T cells. J. Immunol. 2003, 171, 6414–6420. [Google Scholar] [CrossRef] [Green Version]
- Morales-Garcia, J.A.; Echeverry-Alzate, V.; Alonso-Gil, S.; Sanz-SanCristobal, M.; Lopez-Moreno, J.A.; Gil, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase7 inhibition activates adult neurogenesis in hippocampus and subventricular zone in vitro and in vivo. Stem. Cells 2017, 35, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.; Zarruk, J.G.; Ceballos, P.; Pérez, D.I.; Pérez, C.; Perez-Castillo, A.; Moro, M.A.; Brea, J.; Val, C.; Cadavid, M.I.; et al. Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur. J. Med. Chem. 2012, 47, 175–185. [Google Scholar] [CrossRef]
- Kobayashi, T.; Gamanuma, M.; Sasaki, T.; Yamashita, Y.; Yuasa, K.; Kotera, J.; Omori, K. Molecular comparison of rat cyclic nucleotide phosphodiesterase 8 family: Unique expression of PDE8B in rat brain. Gene 2003, 319, 21–31. [Google Scholar] [CrossRef]
- Dong, H.; Osmanova, V.; Epstein, P.M.; Brocke, S. Phosphodiesterase 8 (PDE8) regulates chemotaxis of activated lymphocytes. Biochem. Biophys. Res. Commun. 2006, 345, 713–719. [Google Scholar] [CrossRef]
- Vang, A.G.; Ben-Sasson, S.Z.; Dong, H.; Kream, B.; DeNinno, M.P.; Claffey, M.M.; Housley, W.; Clark, R.B.; Epstein, P.M.; Brocke, S. PDE8 regulates rapid Teff cell adhesion and proliferation independent of ICER. PLoS ONE 2010, 5, e12011. [Google Scholar] [CrossRef] [Green Version]
- Vang, A.G.; Housley, W.; Dong, H.; Basole, C.; Ben-Sasson, S.Z.; Kream, B.E.; Epstein, P.M.; Clark, R.B.; Brocke, S. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: A potential role for Epac. Biochem. J. 2013, 456, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Basole, C.P.; Nguyen, R.K.; Lamothe, K.; Vang, A.; Clark, R.; Baillie, G.S.; Epstein, P.M.; Brocke, S. PDE8 controls CD4+ T cell motility through the PDE8A-Raf-1 kinase signaling complex. Cell Signal 2017, 40, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Vang, A.G.; Basole, C.; Dong, H.; Nguyen, R.K.; Housley, W.; Guernsey, L.; Adami, A.J.; Thrall, R.S.; Clark, R.B.; Epstein, P.M.; et al. Differential expression and function of PDE8 and PDE4 in effector T cells: Implications for PDE8 as a drug target in inflammation. Front. Pharmacol. 2016, 7, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Lerner, A.; Epstein, P.M. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem. J. 2006, 393, 21–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, R.; Wilkinson, I.R.; McCallum, J.F.; Engels, P.; Houslay, M.D. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: Generation of a molecular model. Biochem. J. 1998, 333 Pt 1, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Fréchou, M.; Margaill, I.; Marchand-Leroux, C.; Beray-Berthat, V. Behavioral tests that reveal long-term deficits after permanent focal cerebral ischemia in mouse. Behav. Brain Res. 2019, 360, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Lv, Y.; Wu, X.; Gui, L.; Zhang, Y.; Qiu, J.; Song, G.; Yao, W.; Wan, L.; et al. Cdh1 overexpression improves emotion and cognitive-related behaviors via regulating hippocampal neuroplasticity in global cerebral ischemia rats. Neurochem. Int. 2019, 124, 225–237. [Google Scholar] [CrossRef]
- Zhang, L.; Schallert, T.; Zhang, Z.G.; Jiang, Q.; Arniego, P.; Li, Q.; Lu, M.; Chopp, M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J. Neurosci. Methods 2002, 117, 207–214. [Google Scholar] [CrossRef]
- Balkaya, M.G.; Trueman, R.C.; Boltze, J.; Corbett, D.; Jolkkonen, J. Behavioral outcome measures to improve experimental stroke research. Behav. Brain Res. 2018, 352, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nampoothiri, S.S.; Potluri, T.; Subramanian, H.; Krishnamurthy, R.G. Rodent gymnastics: Neurobehavioral assays in ischemic stroke. Mol. Neurobiol. 2017, 54, 6750–6761. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Blizzard, K.K.; Zeng, Z.; DeVries, A.C.; Hurn, P.D.; McCullough, L.D. Chronic behavioral testing after focal ischemia in the mouse: Functional recovery and the effects of gender. Exp. Neurol. 2004, 187, 94–104. [Google Scholar] [CrossRef]
- Bouët, V.; Freret, T.; Toutain, J.; Divoux, D.; Boulouard, M.; Schumann-Bard, P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp. Neurol. 2007, 203, 555–567. [Google Scholar] [CrossRef]
- Correia Bacarin, C.; Mori, M.A.; Dias Fiuza Ferreira, E.; Valério Romanini, C.; Weffort de Oliveira, R.M.; Milani, H. Fish oil provides robust and sustained memory recovery after cerebral ischemia: Influence of treatment regimen. Physiol. Behav. 2013, 119, 61–71. [Google Scholar] [CrossRef]
- Freret, T.; Chazalviel, L.; Roussel, S.; Bernaudin, M.; Schumann-Bard, P.; Boulouard, M. Long-term functional outcome following transient middle cerebral artery occlusion in the rat: Correlation between brain damage and behavioral impairment. Behav. Neurosci. 2006, 120, 1285–1298. [Google Scholar] [CrossRef]
- Rosell, A.; Agin, V.; Rahman, M.; Morancho, A.; Ali, C.; Koistinaho, J.; Wang, X.; Vivien, D.; Schwaninger, M.; Montaner, J. Distal occlusion of the middle cerebral artery in mice: Are we ready to assess long-term functional outcome? Transl. Stroke Res. 2013, 4, 297–307. [Google Scholar] [CrossRef]
- Boltze, J.; Lukomska, B.; Jolkkonen, J. Mesenchymal stromal cells in stroke: Improvement of motor recovery or functional compensation? J. Cereb. Blood Flow Metab. 2014, 34, 1420–1421. [Google Scholar] [CrossRef]
- Yonemori, F.; Yamaguchi, T.; Yamada, H.; Tamura, A. Spatial cognitive performance after chronic focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 1999, 19, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, C.M.; Oliva, A.A., Jr.; Alonso, O.F.; Pearse, D.D.; Bramlett, H.M.; Dietrich, W.D. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 2007, 208, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.M.; Titus, D.J.; Oliva, A.A., Jr.; Furones, C.; Atkins, C.M. Traumatic brain injury upregulates phosphodiesterase expression in the hippocampus. Front. Syst. Neurosci. 2016, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, G.; Tonon, A.C.; Guella, F.L.; Pettenuzzo, L.F.; Duarte, T.; Duarte, M.; Oses, J.P.; Achaval, M.; Souza, D.O. Guanosine protects against cortical focal ischemia. Involvement of inflammatory response. Mol. Neurobiol. 2015, 52, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, J.; Heckman, P.R.A.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yang, X.M.; Zhuo, Y.Y.; Zhou, H.; Lin, H.B.; Cheng, Y.F.; Xu, J.P.; Zhang, H.T. The phosphodiesterase-4 inhibitor rolipram reverses Aβ-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int. J. Neuropsychopharmacol. 2012, 15, 749–766. [Google Scholar] [CrossRef] [Green Version]
- Manwani, B.; Liu, F.; Xu, Y.; Persky, R.; Li, J.; McCullough, L.D. Functional recovery in aging mice after experimental stroke. Brain Behav. Immun. 2011, 25, 1689–1700. [Google Scholar] [CrossRef] [Green Version]
- Lubjuhn, J.; Gastens, A.; von Wilpert, G.; Bargiotas, P.; Herrmann, O.; Murikinati, S.; Rabie, T.; Marti, H.H.; Amende, I.; Hampton, T.G.; et al. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J. Neurosci. Methods 2009, 184, 95–103. [Google Scholar] [CrossRef]
- Schepers, M.; Tiane, A.; Paes, D.; Sanchez, S.; Rombaut, B.; Piccart, E.; Rutten, B.P.F.; Brône, B.; Hellings, N.; Prickaerts, J.; et al. Targeting phosphodiesterases-towards a tailor-made approach in multiple sclerosis treatment. Front. Immunol. 2019, 10, 1727. [Google Scholar] [CrossRef]
- Wilson, N.M.; Gurney, M.E.; Dietrich, W.D.; Atkins, C.M. Therapeutic benefits of phosphodiesterase 4B inhibition after traumatic brain injury. PLoS ONE 2017, 12, e0178013. [Google Scholar] [CrossRef]
- Titus, D.J.; Wilson, N.M.; Freund, J.E.; Carballosa, M.M.; Sikah, K.E.; Furones, C.; Dietrich, W.D.; Gurney, M.E.; Atkins, C.M. Chronic cognitive dysfunction after traumatic brain injury is improved with a phosphodiesterase 4B inhibitor. J. Neurosci. 2016, 36, 7095–7108. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, G.; Morissette, C.; Lagacé, C.; Boulé, M.; Ouellette, M.J.; McLaughlin, R.W.; Lacombe, D.; Gervais, F.; Tremblay, P. The cAMP-specific phosphodiesterase 4B mediates Abeta-induced microglial activation. Neurobiol. Aging 2006, 27, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cheng, Y.; Wang, C.; Wu, J.; Zou, Z.; Niu, B.; Yu, H.; Wang, H.; Xu, J. FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology 2017, 116, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Rombaut, B.; Kessels, S.; Schepers, M.; Tiane, A.; Paes, D.; Solomina, Y.; Piccart, E.; van den Hove, D.; Brône, B.; Prickaerts, J.; et al. PDE inhibition in distinct cell types to reclaim the balance of synaptic plasticity. Theranostics 2021, 11, 2080–2097. [Google Scholar] [CrossRef] [PubMed]
- González-García, C.; Bravo, B.; Ballester, A.; Gómez-Pérez, R.; Eguiluz, C.; Redondo, M.; Martínez, A.; Gil, C.; Ballester, S. Comparative assessment of PDE 4 and 7 inhibitors as therapeutic agents in experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2013, 170, 602–613. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Study | Compound | Concentration Inhibitor | Administration Route | Time Point of Treatment | Stroke Model | Mouse vs. Rat | Results |
|---|---|---|---|---|---|---|---|
| Kraft et al., 2013 [73] | Rolipram |
| I.P. injection | 2 h post-stroke induction | tMCAO | Male C57Bl/6 mice |
|
| Yang et al., 2014 [74] | Rolipram | 3 mg/kg | I.P. injection | 30 min prior to stroke onset (1) 60 min prior to stroke onset (2) | (1) ligation model (2) embolic model |
| ↑ lesion size |
| Xu et al., 2021 [75] | Roflumilast |
| 2 h post-stroke induction | tMCAO | Male Sprague-Dawley rats |
| |
| Xu et al., 2019 [76] | FCPR03 |
| 2 h post-stroke induction | tMCAO | Male Sprague-Dawley rats |
| |
| Chen et al., 2018 [77] | FCPR16 |
| I.P. injection | 2 h after ischemia | tMCAO |
|
|
| Bonato et al., 2021 [94] | Roflumilast |
| I.P injection | 1 h after reperfusion (continued for 21 days) | Transient global cerebral ischemia | Male Wistar rats |
|
| Soares et al., 2016 [95] | Rolipram |
| I.P. injection | 1 h after reperfusion (continued for 21 days) | Transient global cerebral ischemia | C57Bl/6 mice | Improved functional recovery (EZM, OLT, FST) |
| Behavior | Behavioral Test | Stroke Model Applicability | Mice vs. Rats |
|---|---|---|---|
| Cognition (spatial learning) | Morris water maze | MCAO | Mice and rats [127,134] |
| Cognition (emotional memory and learning) | Fear-conditioning | MCAO | Mice and rats [129,135,136] |
| Cognition (memory) | Passive avoidance | MCAO | Mice and rats [127,129] |
| Cognition (spatial memory) | Aversive eight-arm radial maze | MCAO | Rats [55,130] |
| Sensorimotor function | Cylinder test | MCAO | Mice and rats [127,137] |
| Sensorimotor function | Adhesive removal test | MCAO | Mice and rats [127,131] |
| Sensorimotor function | Rotarod | MCAO (not in the photothrombotic model) | Mice and rats [129,138,139] |
| Sensorimotor function | Grip strength | MCAO | Mice and rats [127,132] |
| Sensorimotor function | Open field test | MCAO | Mice and rats [127,140] |
| Sensorimotor function | Corner test | MCAO | Mice and rats [127,141] |
| Motor function | Staircase test | MCAO | Mice and rats [129,137,142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponsaerts, L.; Alders, L.; Schepers, M.; de Oliveira, R.M.W.; Prickaerts, J.; Vanmierlo, T.; Bronckaers, A. Neuroinflammation in Ischemic Stroke: Inhibition of cAMP-Specific Phosphodiesterases (PDEs) to the Rescue. Biomedicines 2021, 9, 703. https://doi.org/10.3390/biomedicines9070703
Ponsaerts L, Alders L, Schepers M, de Oliveira RMW, Prickaerts J, Vanmierlo T, Bronckaers A. Neuroinflammation in Ischemic Stroke: Inhibition of cAMP-Specific Phosphodiesterases (PDEs) to the Rescue. Biomedicines. 2021; 9(7):703. https://doi.org/10.3390/biomedicines9070703
Chicago/Turabian StylePonsaerts, Laura, Lotte Alders, Melissa Schepers, Rúbia Maria Weffort de Oliveira, Jos Prickaerts, Tim Vanmierlo, and Annelies Bronckaers. 2021. "Neuroinflammation in Ischemic Stroke: Inhibition of cAMP-Specific Phosphodiesterases (PDEs) to the Rescue" Biomedicines 9, no. 7: 703. https://doi.org/10.3390/biomedicines9070703