
Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
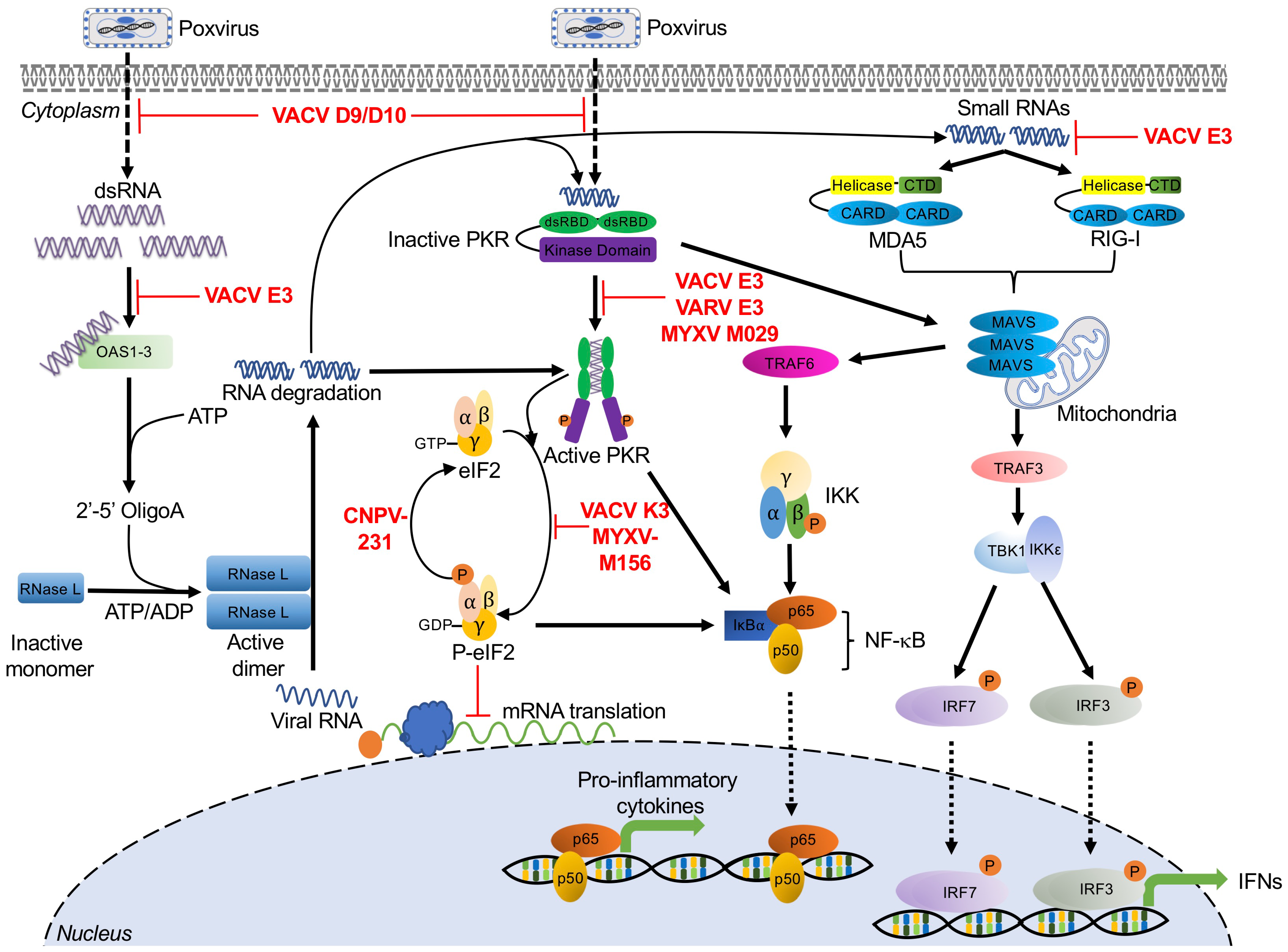
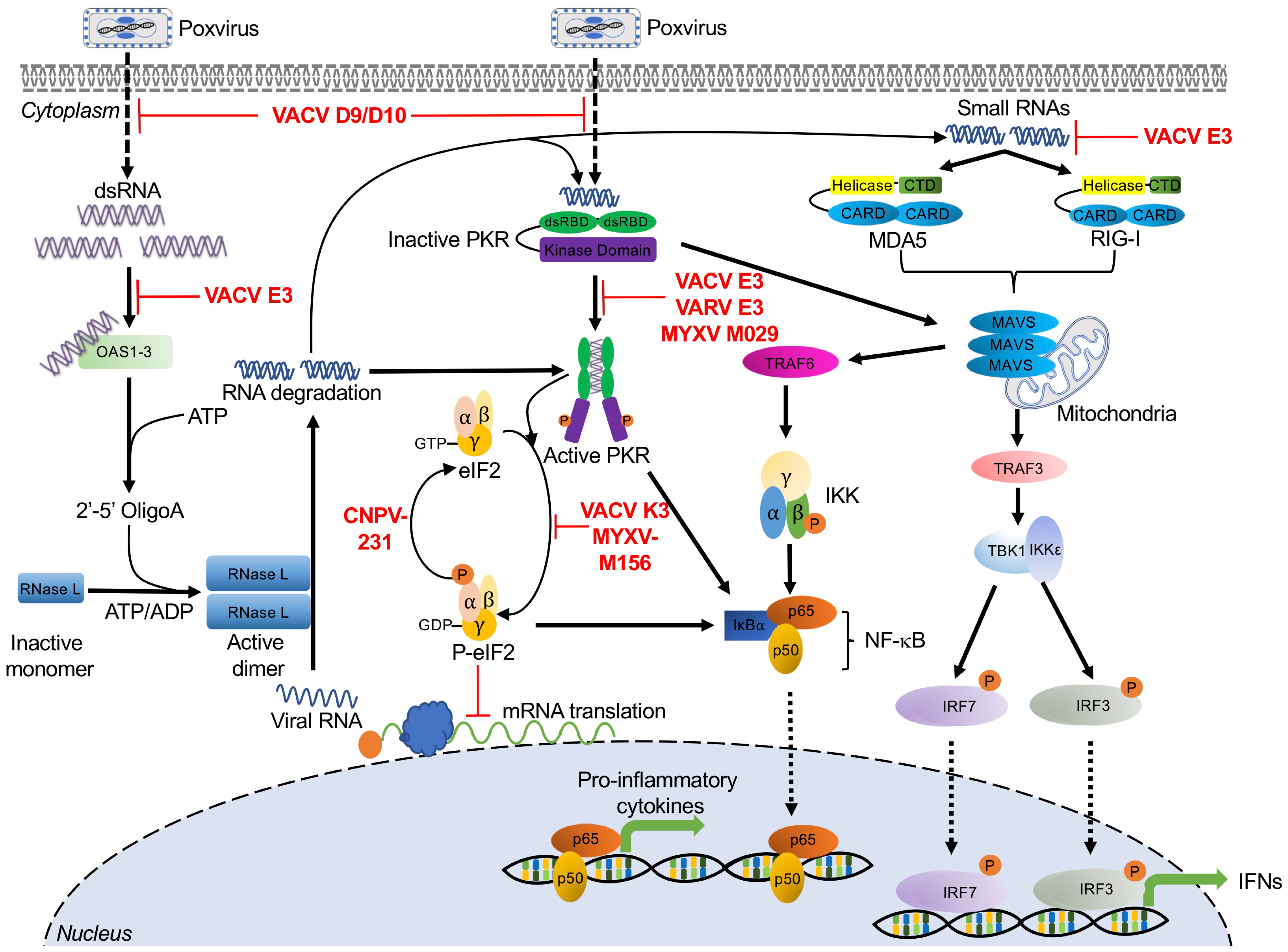
2. Double-Stranded RNA-Activated Sensors and Poxvirus Antagonists
2.1. Protein Kinase R
Poxvirus Evasion of PKR
2.2. 2′-5′-Oligoadenylate Synthetase (OAS)/RNase L
Poxvirus Evasion of OAS/RNase L
2.3. RIG-I and MDA5
Poxvirus Evasion of RIG-I and MDA5
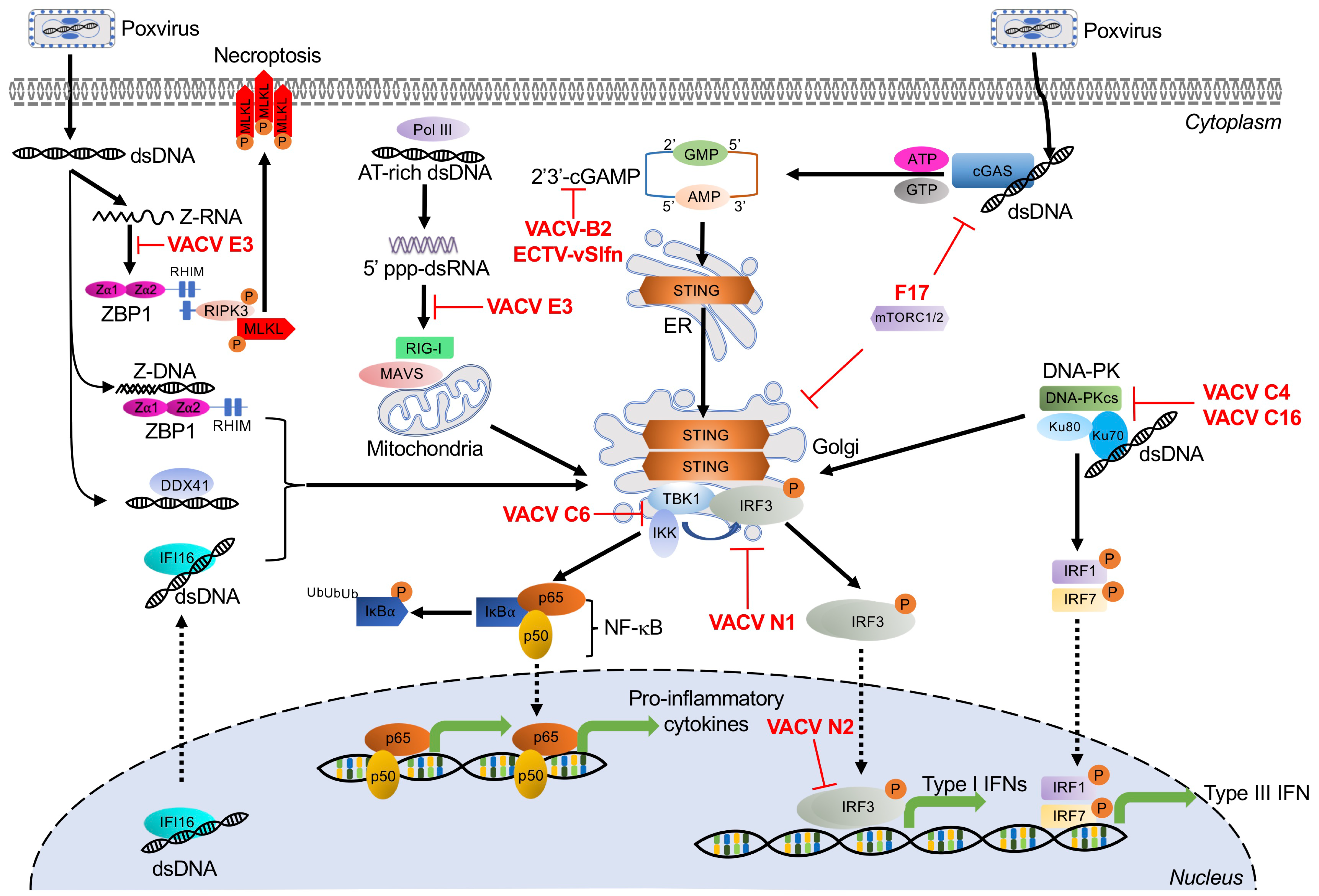
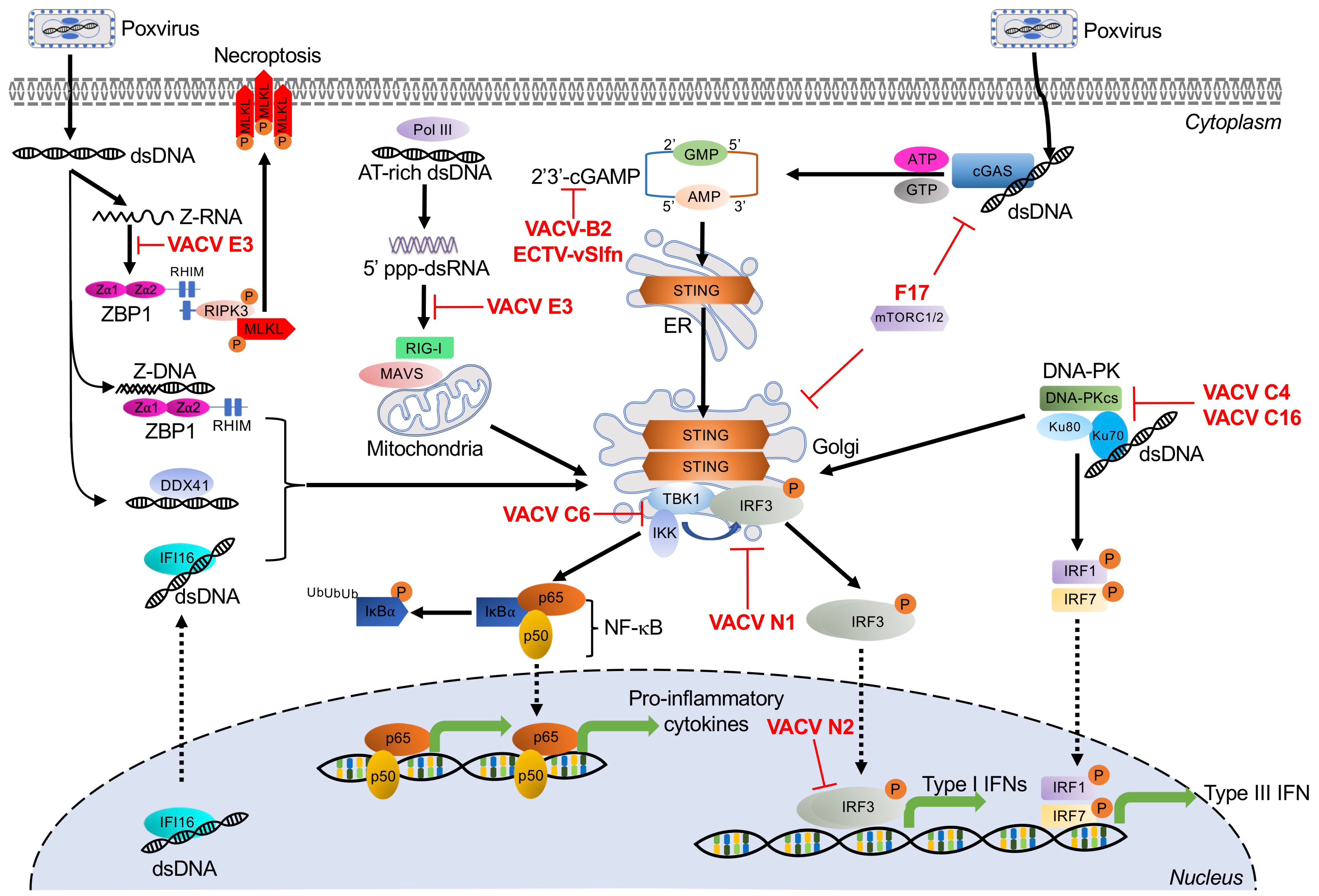
3. Dual RNA/DNA Sensor
Z-DNA Binding Protein 1
Poxvirus Evasion of ZBP1
4. DNA-Activated Sensors and Poxvirus Antagonists
4.1. Cyclic GMP-AMP Synthase
4.1.1. Poxvirus Evasion of cGAS
4.1.2. Poxvirus Evasion of STING
4.2. RNA Polymerase III
Poxvirus Evasion of Pol III
4.3. Interferon-γ Inducible Protein 16
4.4. DNA-Dependent Protein Kinase
Poxvirus Evasion of DNA-PK
4.5. DEAD Box Polypeptide 41
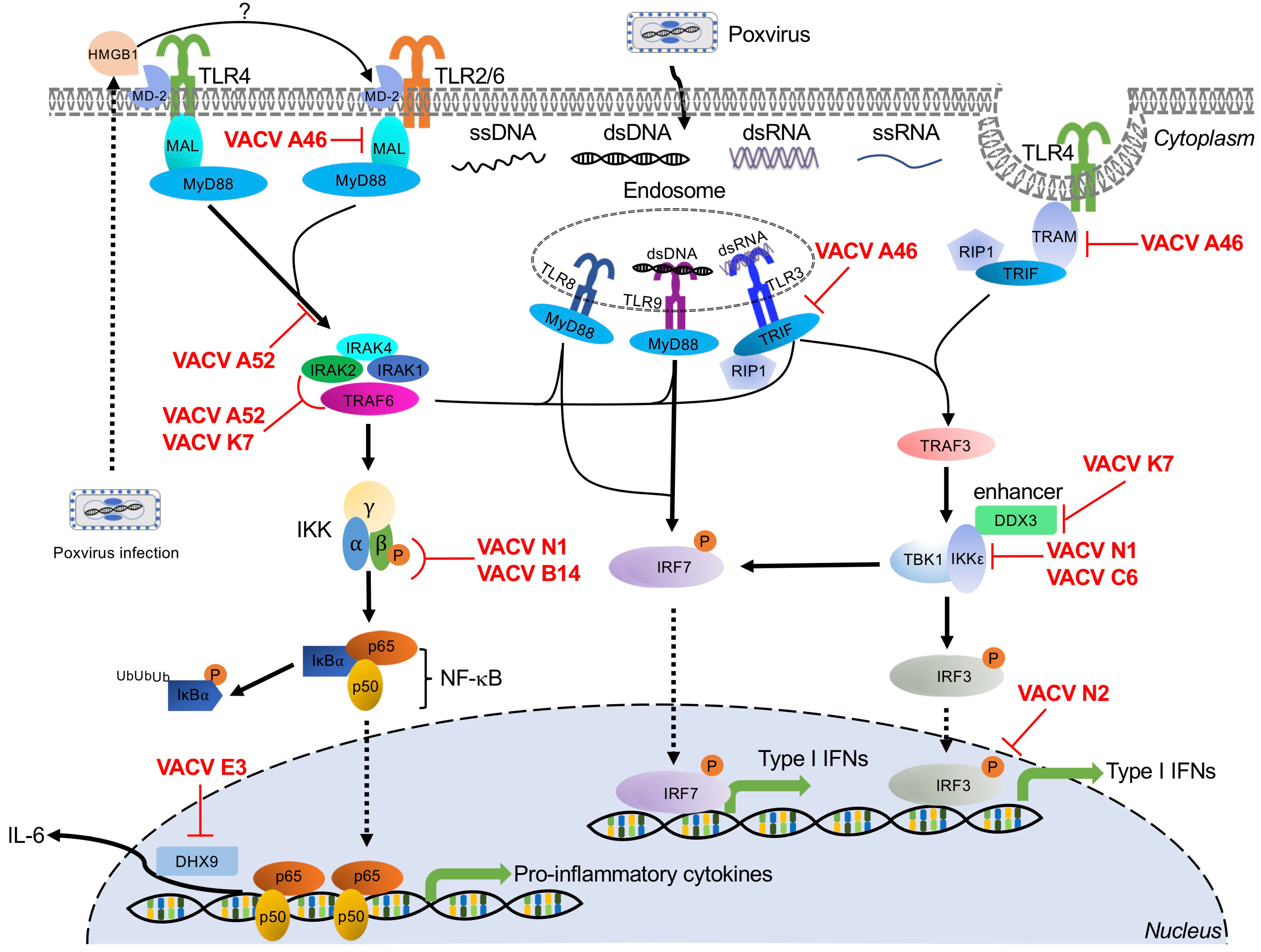
5. Toll-Like Receptor-Mediated Poxvirus Recognition and Poxvirus Antagonists
5.1. TLR3
Poxvirus Evasion of TLR3
5.2. TLR8
Poxvirus Evasion of TLR8
5.3. TLR9
Poxvirus Evasion of TLR9
5.4. TLR4
Poxvirus Evasion of TLR4
5.5. TLR2
Poxvirus Evasion of TLR2
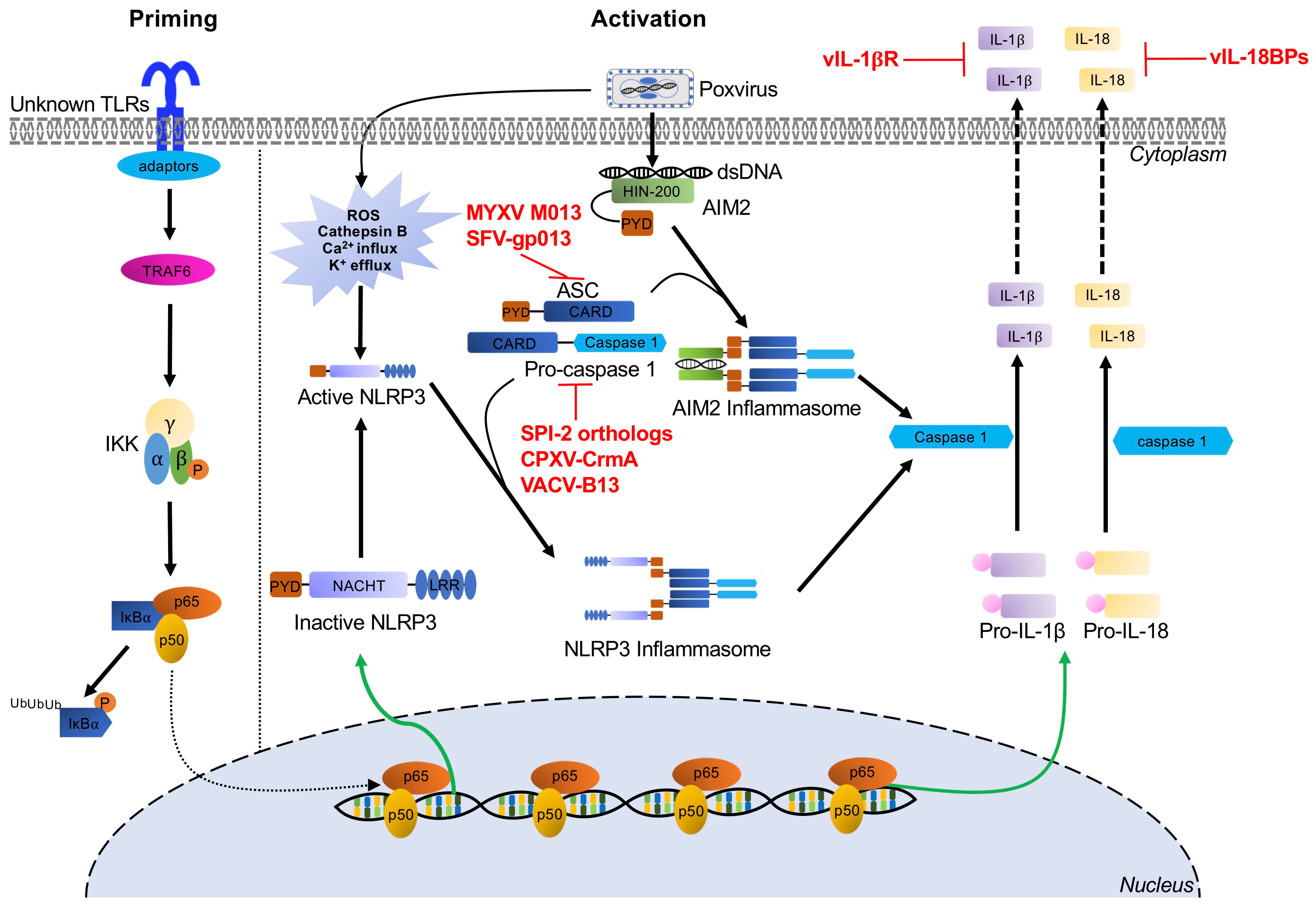
6. Inflammasome Recognition of Poxviruses and Poxvirus Antagonists
6.1. The NLRP3 Inflammasome
Poxvirus Evasion of the NLRP3 Inflammasome
6.2. The AIM2 Inflammasome
7. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 21, 15–40. [Google Scholar] [CrossRef] [Green Version]
- Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.D.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Durski, K.N.; McCollum, A.M.; Nakazawa, Y.; Petersen, B.W.; Reynolds, M.G.; Briand, S.; Khalakdina, A. Emergence of monkeypox in West Africa and Central Africa, 1970–2017. Relev. Epidemiol. Hebd. 2018, 93, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.M.; Venter, E.H.; Shisler, J.L.; Gari, G.; Mekonnen, G.A.; Juleff, N.; Lyons, N.A.; De Clercq, K.; Upton, C.; Bowden, T.R.; et al. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound. Emerg. Dis. 2017, 64, 729–745. [Google Scholar] [CrossRef]
- Spiesschaert, B.; McFadden, G.; Hermans, K.; Nauwynck, H.; Van De Walle, G.R. The current status and future directions of myxoma virus, a master in immune evasion. Vet. Res. 2011, 42, 76. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.L.; Benfield, C.T.O.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Albarnaz, J.D.; Torres, A.A.; Smith, G.L. Modulating Vaccinia Virus Immunomodulators to Improve Immunological Memory. Viruses 2018, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Staib, C.; Kisling, S.; Erfle, V.; Sutter, G. Inactivation of the viral interleukin 1beta receptor improves CD8+ T-cell memory responses elicited upon immunization with modified vaccinia virus Ankara. J. Gen. Virol. 2005, 86, 1997–2006. [Google Scholar] [CrossRef]
- Falivene, J.; Del Médico Zajac, M.P.; Pascutti, M.F.; Rodríguez, A.M.; Maeto, C.; Perdiguero, B.; Gómez, C.E.; Esteban, M.; Calamante, G.; Gherardi, M.M. Improving the MVA vaccine potential by deleting the viral gene coding for the IL-18 binding protein. PLoS ONE 2012, 7, e32220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.L.; Moss, B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene 1983, 25, 21–28. [Google Scholar] [CrossRef]
- Liu, R.; Americo, J.L.; Cotter, C.A.; Earl, P.L.; Erez, N.; Peng, C.; Moss, B. One or two injections of MVA-vectored vaccine shields hACE2 transgenic mice from SARS-CoV-2 upper and lower respiratory tract infection. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Xiao, W.; Americo, J.L.; Earl, P.L.; Moss, B. Novel Nonreplicating Vaccinia Virus Vector Enhances Expression of Heterologous Genes and Suppresses Synthesis of Endogenous Viral Proteins. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef] [Green Version]
- Werden, S.J.; McFadden, G. The role of cell signaling in poxvirus tropism: The case of the M-T5 host range protein of myxoma virus. Biochim. Biophys. Acta 2008, 1784, 228–237. [Google Scholar] [CrossRef]
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro. Oncol. 2016, 18, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Barrett, J.W.; Nazarian, S.H.; Goodwin, M.; Ricciuto, D.; Wang, G.; McFadden, G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 2005, 23, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Urbasic, A.S.; Hynes, S.; Somrak, A.; Contakos, S.; Rahman, M.M.; Liu, J.; MacNeill, A.L. Oncolysis of canine tumor cells by myxoma virus lacking the serp2 gene. Am. J. Vet. Res. 2012, 73, 1252–1261. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Oncolytic Virotherapy with Myxoma Virus. J. Clin. Med. 2020, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Burshtyn, D.N. NK cells and poxvirus infection. Front. Immunol. 2013, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Medeiros-Silva, D.C.; Dos Santos Moreira-Silva, E.A.; de Assis Silva Gomes, J.; da Fonseca, F.G.; Correa-Oliveira, R. CD4 and CD8 T cells participate in the immune memory response against Vaccinia virus after a previous natural infection. Results Immunol. 2013, 3, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pütz, M.M.; Midgley, C.M.; Law, M.; Smith, G.L. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 2006, 12, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Martinez, J.; Huang, X.; Yang, Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 2007, 109, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Hutchens, M.; Luker, K.E.; Sottile, P.; Sonstein, J.; Lukacs, N.W.; Núñez, G.; Curtis, J.L.; Luker, G.D. TLR3 increases disease morbidity and mortality from vaccinia infection. J. Immunol. 2008, 180, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Samuelsson, C.; Hausmann, J.; Lauterbach, H.; Schmidt, M.; Akira, S.; Wagner, H.; Chaplin, P.; Suter, M.; O’Keeffe, M.; Hochrein, H. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J. Clin. Investig. 2008, 118, 1776–1784. [Google Scholar] [CrossRef]
- Wolferstätter, M.; Schweneker, M.; Späth, M.; Lukassen, S.; Klingenberg, M.; Brinkmann, K.; Wielert, U.; Lauterbach, H.; Hochrein, H.; Chaplin, P.; et al. Recombinant modified vaccinia virus Ankara generating excess early double-stranded RNA transiently activates protein kinase R and triggers enhanced innate immune responses. J. Virol. 2014, 88, 14396–14411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.D.; Turner, P.C.; Embury, J.E.; Moldawer, L.L.; Baker, H.V.; Moyer, R.W. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J. Virol. 2011, 85, 550–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.-G.; Le Roy, D.; Knaup Reymond, M.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- El-Jesr, M.; Teir, M.; Maluquer de Motes, C. Vaccinia Virus Activation and Antagonism of Cytosolic DNA Sensing. Front. Immunol. 2020, 11, 568412. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; McFadden, G. Poxvirus immunomodulatory strategies: Current perspectives. J. Virol. 2003, 77, 6093–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: The 2′-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef]
- Tuazon Kels, M.J.; Ng, E.; Al Rumaih, Z.; Pandey, P.; Ruuls, S.R.; Korner, H.; Newsome, T.P.; Chaudhri, G.; Karupiah, G. TNF deficiency dysregulates inflammatory cytokine production, leading to lung pathology and death during respiratory poxvirus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 15935–15946. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Z.; Fuhlbrigge, R.C.; Peña-Cruz, V.; Lieberman, J.; Kupper, T.S. Vaccinia virus induces strong immunoregulatory cytokine production in healthy human epidermal keratinocytes: A novel strategy for immune evasion. J. Virol. 2005, 79, 7363–7370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Piras, V.; Selvarajoo, K. Beyond MyD88 and TRIF Pathways in Toll-Like Receptor Signaling. Front. Immunol. 2014, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Itav, S.; Elinav, E. Integration of Innate Immune Signaling. Trends Immunol. 2016, 37, 84–101. [Google Scholar] [CrossRef]
- Boone, R.F.; Parr, R.P.; Moss, B. Intermolecular duplexes formed from polyadenylylated vaccinia virus RNA. J. Virol. 1979, 30, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Colby, C.; Jurale, C.; Kates, J.R. Mechanism of synthesis of vaccinia virus double-stranded ribonucleic acid in vivo and in vitro. J. Virol. 1971, 7, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Willis, K.L.; Langland, J.O.; Shisler, J.L. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-κB activation, when the K1 protein is absent or mutated. J. Biol. Chem. 2011, 286, 7765–7778. [Google Scholar] [CrossRef] [Green Version]
- Lebleu, B.; Sen, G.C.; Shaila, S.; Cabrer, B.; Lengyel, P. Interferon, double-stranded RNA, and protein phosphorylation. Proc. Natl. Acad. Sci. USA 1976, 73, 3107–3111. [Google Scholar] [CrossRef] [Green Version]
- Hovanessian, A.G.; Brown, R.E.; Kerr, I.M. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 1977, 268, 537–540. [Google Scholar] [CrossRef]
- Kerr, I.M.; Brown, R.E.; Hovanessian, A.G. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature 1977, 268, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.B.; Kerr, I.M.; Williams, B.R.G.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Rebouillat, D.; Hovanessian, A.G. The human 2′,5′-oligoadenylate synthetase family: Interferon-induced proteins with unique enzymatic properties. J. Interf. Cytokine Res. 1999, 19, 295–308. [Google Scholar] [CrossRef]
- Nanduri, S.; Carpick, B.W.; Yang, Y.; Williams, B.R.; Qin, J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998, 17, 5458–5465. [Google Scholar] [CrossRef]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Dever, T.E.; Kinzy, T.G.; Pavitt, G.D. Mechanism and Regulation of Protein Synthesis in Saccharomyces cerevisiae. Genetics 2016, 203, 65–107. [Google Scholar] [CrossRef] [Green Version]
- Adomavicius, T.; Guaita, M.; Zhou, Y.; Jennings, M.D.; Latif, Z.; Roseman, A.M.; Pavitt, G.D. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 2019, 10, 2136. [Google Scholar] [CrossRef] [Green Version]
- Dey, M.; Mann, B.R.; Anshu, A.; Mannan, M.A. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J. Biol. Chem. 2014, 289, 5747–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, C.S.; Taghavi, N.; Samuel, C.E. Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J. Biol. Chem. 2012, 287, 36384–36392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Der, S.D.; Lau, A.S. Involvement of the double-stranded-RNA-dependent kinase PKR in interferon expression and interferon-mediated antiviral activity. Proc. Natl. Acad. Sci. USA 1995, 92, 8841–8845. [Google Scholar] [CrossRef] [Green Version]
- Gil, J.; Rullas, J.; García, M.A.; Alcamí, J.; Esteban, M. The catalytic activity of dsRNA-dependent protein kinase, PKR, is required for NF-kappaB activation. Oncogene 2001, 20, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Pham, A.M.; Santa Maria, F.G.; Lahiri, T.; Friedman, E.; Marié, I.J.; Levy, D.E. PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLoS Pathog. 2016, 12, e1005489. [Google Scholar] [CrossRef]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2α. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Jacobs, B.L. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 1993, 194, 537–547. [Google Scholar] [CrossRef]
- Romano, P.R.; Zhang, F.; Tan, S.-L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of Double-Stranded RNA-Dependent Protein Kinase PKR by Vaccinia Virus E3: Role of Complex Formation and the E3 N-Terminal Domain. Mol. Cell. Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef] [Green Version]
- Myskiw, C.; Arsenio, J.; Hammett, C.; van Bruggen, R.; Deschambault, Y.; Beausoleil, N.; Babiuk, S.; Cao, J. Comparative analysis of poxvirus orthologues of the vaccinia virus E3 protein: Modulation of protein kinase R activity, cytokine responses, and virus pathogenicity. J. Virol. 2011, 85, 12280–12291. [Google Scholar] [CrossRef] [Green Version]
- Langland, J.O.; Jacobs, B.L. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 2002, 299, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Jacobs, B.L.; Samuel, C.E. Loss of Protein Kinase PKR Expression in Human HeLa Cells Complements the Vaccinia Virus E3L Deletion Mutant Phenotype by Restoration of Viral Protein Synthesis. J. Virol. 2008, 82, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef]
- Park, C.; Peng, C.; Brennan, G.; Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Ann. N. Y. Acad. Sci. 2019, 1438, 18–29. [Google Scholar] [CrossRef]
- Beattie, E.; Tartaglia, J.; Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology 1991, 183, 419–422. [Google Scholar] [CrossRef]
- Dar, A.C.; Sicheri, F. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol. Cell 2002, 10, 295–305. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Cort, J.R.; Yee, A.A.; Liu, F.; Goshe, M.B.; Edwards, A.M.; Smith, R.D.; Arrowsmith, C.H.; Dever, T.E.; Kennedy, M.A. Myxoma virus immunomodulatory protein M156R is a structural mimic of eukaryotic translation initiation factor eIF2alpha. J. Mol. Biol. 2002, 322, 943–954. [Google Scholar] [CrossRef]
- Rothenburg, S.; Seo, E.J.; Gibbs, J.S.; Dever, T.E.; Dittmar, K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009, 16, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Haller, S.L.; Rahman, M.M.; McFadden, G.; Rothenburg, S. Myxoma virus M156 is a specific inhibitor of rabbit PKR but contains a loss-of-function mutation in Australian virus isolates. Proc. Natl. Acad. Sci. USA 2016, 113, 3855–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Peng, C.; Rahman, M.J.; Haller, S.L.; Tazi, L.; Brennan, G.; Rothenburg, S. Orthopoxvirus K3 orthologs show virus- and host-specific inhibition of the antiviral protein kinase PKR. PLoS Pathog. 2021, 17, e1009183. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Varga, J.; Deschambault, Y. Poxvirus encoded eIF2α homolog, K3 family proteins, is a key determinant of poxvirus host species specificity. Virology 2020, 541, 101–112. [Google Scholar] [CrossRef]
- Shors, T.; Keck, J.G.; Moss, B. Down regulation of gene expression by the vaccinia virus D10 protein. J. Virol. 1999, 73, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Parrish, S.; Moss, B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007, 81, 12973–12978. [Google Scholar] [CrossRef] [Green Version]
- Parrish, S.; Resch, W.; Moss, B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. USA 2007, 104, 2139–2144. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-W.; Katsafanas, G.C.; Liu, R.; Wyatt, L.S.; Moss, B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe 2015, 17, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Burgess, H.M.; Mohr, I. Cellular 5′-3′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 2015, 17, 332–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratke, K.A.; McLysaght, A.; Rothenburg, S. A survey of host range genes in poxvirus genomes. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2013, 14, 406–425. [Google Scholar] [CrossRef] [Green Version]
- Chebath, J.; Benech, P.; Hovanessian, A.; Galabru, J.; Revel, M. Four different forms of interferon-induced 2′,5′-oligo(A) synthetase identified by immunoblotting in human cells. J. Biol. Chem. 1987, 262, 3852–3857. [Google Scholar] [CrossRef]
- Silverman, R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dong, B.; Wei, Z.; Silverman, R.H.; Weiss, S.R. Activation of RNase L in Egyptian Rousette Bat-Derived RoNi/7 Cells Is Dependent Primarily on OAS3 and Independent of MAVS Signaling. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Luthra, P.; Sun, D.; Silverman, R.H.; He, B. Activation of IFN-β expression by a viral mRNA through RNase L and MDA5. Proc. Natl. Acad. Sci. USA 2011, 108, 2118–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malathi, K.; Dong, B.; Gale, M.J.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [Green Version]
- Rong, E.; Wang, X.; Chen, H.; Yang, C.; Hu, J.; Liu, W.; Wang, Z.; Chen, X.; Zheng, H.; Pu, J.; et al. Molecular Mechanisms for the Adaptive Switching Between the OAS/RNase L and OASL/RIG-I Pathways in Birds and Mammals. Front. Immunol. 2018, 9, 1398. [Google Scholar] [CrossRef]
- Manivannan, P.; Siddiqui, M.A.; Malathi, K. RNase L Amplifies Interferon Signaling by Inducing Protein Kinase R-Mediated Antiviral Stress Granules. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Guerra, M.; Rivas, C.; Esteban, M. Inducible expression of the 2-5A synthetase/RNase L system results in inhibition of vaccinia virus replication. Virology 1997, 227, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Rivas, C.; Gil, J.; Mĕlková, Z.; Esteban, M.; Díaz-Guerra, M. Vaccinia virus E3L protein is an inhibitor of the interferon (i.f.n.)-induced 2-5A synthetase enzyme. Virology 1998, 243, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Mages, J.; Staib, C.; Lehmann, M.H.; Lang, R.; Sutter, G. Role of viral factor E3L in modified vaccinia virus ankara infection of human HeLa Cells: Regulation of the virus life cycle and identification of differentially expressed host genes. J. Virol. 2005, 79, 2584–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Condit, R.C.; Vijaysri, S.; Jacobs, B.; Williams, B.R.G.; Silverman, R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002, 76, 5251–5259. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-W.; Wyatt, L.S.; Orandle, M.S.; Minai, M.; Moss, B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J. Virol. 2014, 88, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Moss, B. Opposing Roles of Double-Stranded RNA Effector Pathways and Viral Defense Proteins Revealed with CRISPR-Cas9 Knockout Cell Lines and Vaccinia Virus Mutants. J. Virol. 2016, 90, 7864–7879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Olagnier, D.; Lin, R. Host and Viral Modulation of RIG-I-Mediated Antiviral Immunity. Front. Immunol. 2016, 7, 662. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gao, X.; Barrett, J.W.; Shao, Q.; Bartee, E.; Mohamed, M.R.; Rahman, M.; Werden, S.; Irvine, T.; Cao, J.; et al. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 2008, 4, e1000099. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Dai, P.; Parikh, T.; Cao, H.; Bhoj, V.; Sun, Q.; Chen, Z.; Merghoub, T.; Houghton, A.; Shuman, S. Vaccinia virus subverts a mitochondrial antiviral signaling protein-dependent innate immune response in keratinocytes through its double-stranded RNA binding protein, E3. J. Virol. 2008, 82, 10735–10746. [Google Scholar] [CrossRef] [Green Version]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Myong, S.; Cui, S.; Cornish, P.V.; Kirchhofer, A.; Gack, M.U.; Jung, J.U.; Hopfner, K.-P.; Ha, T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science 2009, 323, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myskiw, C.; Arsenio, J.; Booy, E.P.; Hammett, C.; Deschambault, Y.; Gibson, S.B.; Cao, J. RNA species generated in vaccinia virus infected cells activate cell type-specific MDA5 or RIG-I dependent interferon gene transcription and PKR dependent apoptosis. Virology 2011, 413, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Valentine, R.; Smith, G.L. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J. Gen. Virol. 2010, 91, 2221–2229. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Comella, N.; Tognazzi, K.; Brown, L.F.; Dvorak, H.F.; Kocher, O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 1999, 240, 157–163. [Google Scholar] [CrossRef]
- Schwartz, T.; Behlke, J.; Lowenhaupt, K.; Heinemann, U.; Rich, A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol. 2001, 8, 761–765. [Google Scholar] [CrossRef]
- Rothenburg, S.; Schwartz, T.; Koch-Nolte, F.; Haag, F. Complex regulation of the human gene for the Z-DNA binding protein DLM-1. Nucleic Acids Res. 2002, 30, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Deigendesch, N.; Koch-Nolte, F.; Rothenburg, S. ZBP1 subcellular localization and association with stress granules is controlled by its Z-DNA binding domains. Nucleic Acids Res. 2006, 34, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Li, H.; Zhao, J.; Pervushin, K.; Lowenhaupt, K.; Schwartz, T.U.; Dröge, P. Alternate rRNA secondary structures as regulators of translation. Nat. Struct. Mol. Biol. 2011, 18, 169–176. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [Green Version]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Früh, K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 2010, 84, 585–598. [Google Scholar] [CrossRef] [Green Version]
- Koehler, H.; Cotsmire, S.; Langland, J.; Kibler, K.V.; Kalman, D.; Upton, J.W.; Mocarski, E.S.; Jacobs, B.L. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 2017, 114, 11506–11511. [Google Scholar] [CrossRef] [Green Version]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.-C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, H.S.; Jacobs, B.L. Subversion of Programed Cell Death by Poxviruses. Curr. Top. Microbiol. Immunol. 2021. [Google Scholar] [CrossRef]
- Brandt, T.A.; Jacobs, B.L. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 2001, 75, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-G.; Muralinath, M.; Brandt, T.; Pearcy, M.; Hauns, K.; Lowenhaupt, K.; Jacobs, B.L.; Rich, A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 6974–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006, 7, 40–48. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479–480, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Xu, R.-H.; Wong, E.B.; Rubio, D.; Roscoe, F.; Ma, X.; Nair, S.; Remakus, S.; Schwendener, R.; John, S.; Shlomchik, M.; et al. Sequential Activation of Two Pathogen-Sensing Pathways Required for Type I Interferon Expression and Resistance to an Acute DNA Virus Infection. Immunity 2015, 43, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-Y.; He, X.-B.; Jia, H.-J.; Chen, G.-H.; Jin, Q.-W.; Long, Z.-L.; Jing, Z.-Z. The cGas-Sting Signaling Pathway Is Required for the Innate Immune Response Against Ectromelia Virus. Front. Immunol. 2018, 9, 1297. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Wang, W.; Cao, H.; Avogadri, F.; Dai, L.; Drexler, I.; Joyce, J.A.; Li, X.-D.; Chen, Z.; Merghoub, T.; et al. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog. 2014, 10, e1003989. [Google Scholar] [CrossRef] [Green Version]
- Georgana, I.; Sumner, R.P.; Towers, G.J.; Maluquer de Motes, C. Virulent Poxviruses Inhibit DNA Sensing by Preventing STING Activation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Meade, N.; King, M.; Munger, J.; Walsh, D. mTOR Dysregulation by Vaccinia Virus F17 Controls Multiple Processes with Varying Roles in Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157.e17. [Google Scholar] [CrossRef] [Green Version]
- Eaglesham, J.B.; Pan, Y.; Kupper, T.S.; Kranzusch, P.J. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 2019, 566, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, B.; Alonso, G.; Georgana, I.; El-Jesr, M.; Martín, R.; Shair, K.H.Y.; Fischer, C.; Sauer, S.; Maluquer de Motes, C.; Alcamí, A. Viral cGAMP nuclease reveals the essential role of DNA sensing in protection against acute lethal virus infection. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011, 7, e1002247. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Benfield, C.T.O.; Ren, H.; Lee, V.H.; Frazer, G.L.; Strnadova, P.; Sumner, R.P.; Smith, G.L. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes virulence. J. Gen. Virol. 2013, 94, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, E.P.; Abascal-Palacios, G.; Daiß, J.L.; King, H.; Gouge, J.; Pilsl, M.; Beuron, F.; Morris, E.; Gunkel, P.; Engel, C.; et al. Structure of human RNA polymerase III. Nat. Commun. 2020, 11, 6409. [Google Scholar] [CrossRef]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Stratmann, S.A.; Morrone, S.R.; van Oijen, A.M.; Sohn, J. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. Elife 2015, 4, e11721. [Google Scholar] [CrossRef] [PubMed]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.E.; Ferguson, B.J.; Mazzon, M.; Fahy, A.S.; Krysztofinska, E.; Arribas-Bosacoma, R.; Pearl, L.H.; Ren, H.; Smith, G.L. A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog. 2013, 9, e1003649. [Google Scholar] [CrossRef] [PubMed]
- Scutts, S.R.; Ember, S.W.; Ren, H.; Ye, C.; Lovejoy, C.A.; Mazzon, M.; Veyer, D.L.; Sumner, R.P.; Smith, G.L. DNA-PK Is Targeted by Multiple Vaccinia Virus Proteins to Inhibit DNA Sensing. Cell Rep. 2018, 25, 1953–1965.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, A.S.; Clark, R.H.; Glyde, E.F.; Smith, G.L. Vaccinia virus protein C16 acts intracellularly to modulate the host response and promote virulence. J. Gen. Virol. 2008, 89, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Parvatiyar, K.; Zhang, Z.; Teles, R.M.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Omura, H.; Oikawa, D.; Nakane, T.; Kato, M.; Ishii, R.; Ishitani, R.; Tokunaga, F.; Nureki, O. Structural and Functional Analysis of DDX41: A bispecific immune receptor for DNA and cyclic dinucleotide. Sci. Rep. 2016, 6, 34756. [Google Scholar] [CrossRef] [Green Version]
- Rock, F.L.; Hardiman, G.; Timans, J.C.; Kastelein, R.A.; Bazan, J.F. A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. USA 1998, 95, 588–593. [Google Scholar] [CrossRef] [Green Version]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Michel, T.; Reichhart, J.M.; Hoffmann, J.A.; Royet, J. Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature 2001, 414, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zhang, H.; Zhao, C.; Zhang, H. Evolutionary History of the Toll-Like Receptor Gene Family across Vertebrates. Genome Biol. Evol. 2020, 12, 3615–3634. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The structural biology of Toll-like receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Pierce, S.K. How location governs toll-like receptor signaling. Traffic 2009, 10, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- De Nardo, D.; Balka, K.R.; Cardona Gloria, Y.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J. Biol. Chem. 2018, 293, 15195–15207. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Macartney, T.; Peggie, M.; Cohen, P. Interleukin-1 and TRAF6-dependent activation of TAK1 in the absence of TAB2 and TAB3. Biochem. J. 2017, 474, 2235–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000166. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.H.; Choi, D.-S. Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells 2019, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, K.-N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Israely, T.; Melamed, S.; Achdout, H.; Erez, N.; Politi, B.; Waner, T.; Lustig, S.; Paran, N. TLR3 and TLR9 agonists improve postexposure vaccination efficacy of live smallpox vaccines. PLoS ONE 2014, 9, e110545. [Google Scholar] [CrossRef] [PubMed]
- Harte, M.T.; Haga, I.R.; Maloney, G.; Gray, P.; Reading, P.C.; Bartlett, N.W.; Smith, G.L.; Bowie, A.; O’Neill, L.A.J. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003, 197, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.; Haga, I.R.; Schröder, M.; Bartlett, N.W.; Maloney, G.; Reading, P.C.; Fitzgerald, K.A.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 2005, 201, 1007–1018. [Google Scholar] [CrossRef]
- DiPerna, G.; Stack, J.; Bowie, A.G.; Boyd, A.; Kotwal, G.; Zhang, Z.; Arvikar, S.; Latz, E.; Fitzgerald, K.A.; Marshall, W.L. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 2004, 279, 36570–36578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.A.-J.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.-J.; Whalen, D.M.; Abrescia, N.G.A.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef] [Green Version]
- Schröder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Funami, K.; Tatematsu, M.; Seya, T.; Matsumoto, M. Endosomal localization of TLR8 confers distinctive proteolytic processing on human myeloid cells. J. Immunol. 2014, 193, 5118–5128. [Google Scholar] [CrossRef] [Green Version]
- Ostendorf, T.; Zillinger, T.; Andryka, K.; Schlee-Guimaraes, T.M.; Schmitz, S.; Marx, S.; Bayrak, K.; Linke, R.; Salgert, S.; Wegner, J.; et al. Immune Sensing of Synthetic, Bacterial, and Protozoan RNA by Toll-like Receptor 8 Requires Coordinated Processing by RNase T2 and RNase 2. Immunity 2020, 52, 591–605.e6. [Google Scholar] [CrossRef]
- Greulich, W.; Wagner, M.; Gaidt, M.M.; Stafford, C.; Cheng, Y.; Linder, A.; Carell, T.; Hornung, V. TLR8 Is a Sensor of RNase T2 Degradation Products. Cell 2019, 179, 1264–1275.e13. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Huang, X.; Yang, Y. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc. Natl. Acad. Sci. USA 2010, 107, 6442–6447. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, A.; Keating, S.E.; Carty, M.; Bowie, A.G. Poxviral protein E3-altered cytokine production reveals that DExD/H-box helicase 9 controls Toll-like receptor-stimulated immune responses. J. Biol. Chem. 2018, 293, 14989–15001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Yamamoto, C.; Matsumoto, F.; Onji, M.; Shibata, T.; Murakami, Y.; Kanno, A.; Hayashi, T.; Tanimura, N.; Yoshida, N.; et al. Cleavage of Toll-Like Receptor 9 Ectodomain Is Required for In Vivo Responses to Single Strand DNA. Front. Immunol. 2018, 9, 1491. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Brinkmann, M.M.; Spooner, E.; Lee, C.C.; Kim, Y.-M.; Ploegh, H.L. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 2008, 9, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Uyangaa, E.; Choi, J.Y.; Patil, A.M.; Hossain, F.M.A.; Park, S.O.; Kim, B.; Kim, K.; Eo, S.K. Dual TLR2/9 Recognition of Herpes Simplex Virus Infection Is Required for Recruitment and Activation of Monocytes and NK Cells and Restriction of Viral Dissemination to the Central Nervous System. Front. Immunol. 2018, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Lousberg, E.L.; Diener, K.R.; Fraser, C.K.; Phipps, S.; Foster, P.S.; Chen, W.; Uematsu, S.; Akira, S.; Robertson, S.A.; Brown, M.P.; et al. Antigen-specific T-cell responses to a recombinant fowlpox virus are dependent on MyD88 and interleukin-18 and independent of Toll-like receptor 7 (TLR7)- and TLR9-mediated innate immune recognition. J. Virol. 2011, 85, 3385–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, P.; Cao, H.; Merghoub, T.; Avogadri, F.; Wang, W.; Parikh, T.; Fang, C.-M.; Pitha, P.M.; Fitzgerald, K.A.; Rahman, M.M.; et al. Myxoma virus induces type I interferon production in murine plasmacytoid dendritic cells via a TLR9/MyD88-, IRF5/IRF7-, and IFNAR-dependent pathway. J. Virol. 2011, 85, 10814–10825. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A.J. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Flo, T.H.; Ryan, L.; Latz, E.; Takeuchi, O.; Monks, B.G.; Lien, E.; Halaas, Ø.; Akira, S.; Skjåk-Braek, G.; Golenbock, D.T.; et al. Involvement of toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J. Biol. Chem. 2002, 277, 35489–35495. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Sugawara, S.; Monodane, T.; Nishijima, M.; Adachi, Y.; Akashi, S.; Miyake, K.; Hase, S.; Takada, H. Micrococcus luteus teichuronic acids activate human and murine monocytic cells in a CD14- and toll-like receptor 4-dependent manner. Infect. Immun. 2001, 69, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an Innate Immune Response In vivo via TLR4. Front. Microbiol. 2017, 8, 1571. [Google Scholar] [CrossRef] [Green Version]
- Georgel, P.; Jiang, Z.; Kunz, S.; Janssen, E.; Mols, J.; Hoebe, K.; Bahram, S.; Oldstone, M.B.A.; Beutler, B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 2007, 362, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Sikorski, R.; Kirn, D.H.; Thorne, S.H. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011, 18, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Matsumiya, M.; Stylianou, E.; Griffiths, K.; Lang, Z.; Meyer, J.; Harris, S.A.; Rowland, R.; Minassian, A.M.; Pathan, A.A.; Fletcher, H.; et al. Roles for Treg expansion and HMGB1 signaling through the TLR1-2-6 axis in determining the magnitude of the antigen-specific immune response to MVA85A. PLoS ONE 2013, 8, e67922. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Hutchens, M.A.; Luker, K.E.; Sonstein, J.; Núñez, G.; Curtis, J.L.; Luker, G.D. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathog. 2008, 4, e1000153. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lysakova-Devine, T.; Keogh, B.; Harrington, B.; Nagpal, K.; Halle, A.; Golenbock, D.T.; Monie, T.; Bowie, A.G. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 2010, 185, 4261–4271. [Google Scholar] [CrossRef] [Green Version]
- Fedosyuk, S.; Bezerra, G.A.; Radakovics, K.; Smith, T.K.; Sammito, M.; Bobik, N.; Round, A.; Ten Eyck, L.F.; Djinović-Carugo, K.; Usón, I.; et al. Vaccinia Virus Immunomodulator A46: A Lipid and Protein-Binding Scaffold for Sequestering Host TIR-Domain Proteins. PLoS Pathog. 2016, 12, e1006079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, J.; Bowie, A.G. Poxviral protein A46 antagonizes Toll-like receptor 4 signaling by targeting BB loop motifs in Toll-IL-1 receptor adaptor proteins to disrupt receptor:adaptor interactions. J. Biol. Chem. 2012, 287, 22672–22682. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, H.; Heo, L.; Seok, C.; Choe, J. Structure of vaccinia virus A46, an inhibitor of TLR4 signaling pathway, shows the conformation of VIPER motif. Protein Sci. 2014, 23, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Azar, D.F.; Haas, M.; Fedosyuk, S.; Rahaman, M.H.; Hedger, A.; Kobe, B.; Skern, T. Vaccinia Virus Immunomodulator A46: Destructive Interactions with MAL and MyD88 Shown by Negative-Stain Electron Microscopy. Structure 2020, 28, 1271–1287.e5. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; Kiss-Toth, E.; Symons, J.A.; Smith, G.L.; Dower, S.K.; O’Neill, L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 10162–10167. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.Y.; Nan, X.; Jin, M.S.; Youn, S.-J.; Ryu, Y.H.; Mah, S.; Han, S.H.; Lee, H.; Paik, S.-G.; Lee, J.-O. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009, 31, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, D.; Joo, H.-S.; Franz-Wachtel, M.; Hertlein, T.; Stevanovic, S.; Macek, B.; Wolz, C.; Götz, F.; Otto, M.; Kretschmer, D.; et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat. Commun. 2016, 7, 12304. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, K.; Cardona Gloria, Y.; Wolz, O.-O.; Herster, F.; Sharma, L.; Dillen, C.A.; Täumer, C.; Dickhöfer, S.; Bittner, Z.; Dang, T.-M.; et al. The fungal ligand chitin directly binds TLR2 and triggers inflammation dependent on oligomer size. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Rosenthal, K.L. HIV-1 Structural Proteins Serve as PAMPs for TLR2 Heterodimers Significantly Increasing Infection and Innate Immune Activation. Front. Immunol. 2015, 6, 426. [Google Scholar] [CrossRef]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.-J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- O’Gorman, W.E.; Sampath, P.; Simonds, E.F.; Sikorski, R.; O’Malley, M.; Krutzik, P.O.; Chen, H.; Panchanathan, V.; Chaudhri, G.; Karupiah, G.; et al. Alternate mechanisms of initial pattern recognition drive differential immune responses to related poxviruses. Cell Host Microbe 2010, 8, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Huang, X.; Yang, Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog. 2010, 6, e1000811. [Google Scholar] [CrossRef] [PubMed]
- González, J.M.; Esteban, M. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: Sequence similarity and evolutionary history. Virol. J. 2010, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maluquer de Motes, C.; Cooray, S.; Ren, H.; Almeida, G.M.F.; McGourty, K.; Bahar, M.W.; Stuart, D.I.; Grimes, J.M.; Graham, S.C.; Smith, G.L. Inhibition of apoptosis and NF-κB activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PLoS Pathog. 2011, 7, e1002430. [Google Scholar] [CrossRef] [Green Version]
- Stack, J.; Doyle, S.L.; Connolly, D.J.; Reinert, L.S.; O’Keeffe, K.M.; McLoughlin, R.M.; Paludan, S.R.; Bowie, A.G. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J. Immunol. 2014, 193, 6090–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, G.; León-Juárez, M.; García-Cordero, J.; Meza-Sánchez, D.E.; Cedillo-Barrón, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef] [Green Version]
- Hayward, J.A.; Mathur, A.; Ngo, C.; Man, S.M. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol. Mol. Biol. Rev. 2018, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; de Alba, E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. Int. J. Mol. Sci. 2021, 22, 872. [Google Scholar] [CrossRef] [PubMed]
- Nambayan, R.J.T.; Sandin, S.I.; Quint, D.A.; Satyadi, D.M.; de Alba, E. The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains, PYD and CARD. J. Biol. Chem. 2019, 294, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Myxoma virus lacking the pyrin-like protein M013 is sensed in human myeloid cells by both NLRP3 and multiple Toll-like receptors, which independently activate the inflammasome and NF-κB innate response pathways. J. Virol. 2011, 85, 12505–12517. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, G.E.; Sand, J.; Sauter, M.; Seyffert, M.; Steigerwald, R.; Fraefel, C.; Smola, S.; French, L.E.; Beer, H.-D. IFN-γ Primes Keratinocytes for HSV-1-Induced Inflammasome Activation. J. Investig. Dermatol. 2016, 136, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Dorfleutner, A.; Talbott, S.J.; Bryan, N.B.; Funya, K.N.; Rellick, S.L.; Reed, J.C.; Shi, X.; Rojanasakul, Y.; Flynn, D.C.; Stehlik, C. A Shope Fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes 2007, 35, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Mohamed, M.R.; Kim, M.; Smallwood, S.; McFadden, G. Co-regulation of NF-kappaB and inflammasome-mediated inflammatory responses by myxoma virus pyrin domain-containing protein M013. PLoS Pathog. 2009, 5, e1000635. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.R.; Jackson, C.B.; Rahman, M.M.; Khan, A.R.; Lewin, A.S.; McFadden, G. Myxoma virus M013 protein antagonizes NF-κB and inflammasome pathways via distinct structural motifs. J. Biol. Chem. 2019, 294, 8480–8489. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Howard, S.T.; Chan, Y.S. Vaccinia virus encodes a family of genes with homology to serine proteinase inhibitors. J. Gen. Virol. 1989, 70 Pt 9, 2333–2343. [Google Scholar] [CrossRef]
- Ray, C.A.; Black, R.A.; Kronheim, S.R.; Greenstreet, T.A.; Sleath, P.R.; Salvesen, G.S.; Pickup, D.J. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 1992, 69, 597–604. [Google Scholar] [CrossRef]
- Bloomer, D.T.; Kitevska-Ilioski, T.; Pantaki-Eimany, D.; Ji, Y.; Miles, M.A.; Heras, B.; Hawkins, C.J. CrmA orthologs from diverse poxviruses potently inhibit caspases-1 and -8, yet cleavage site mutagenesis frequently produces caspase-1-specific variants. Biochem. J. 2019, 476, 1335–1357. [Google Scholar] [CrossRef]
- Alcamí, A.; Smith, G.L. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: A novel mechanism of virus modulation of the host response to infection. Cell 1992, 71, 153–167. [Google Scholar] [CrossRef]
- Smith, V.P.; Alcami, A. Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J. Virol. 2000, 74, 8460–8471. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Moss, B. IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.P.; Bryant, N.A.; Alcamí, A. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J. Gen. Virol. 2000, 81, 1223–1230. [Google Scholar] [CrossRef]
- Meng, X.; Leman, M.; Xiang, Y. Variola virus IL-18 binding protein interacts with three human IL-18 residues that are part of a binding site for human IL-18 receptor alpha subunit. Virology 2007, 358, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattgen, S.A.; Fitzgerald, K.A. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 2011, 243, 109–118. [Google Scholar] [CrossRef]
- Lu, A.; Li, Y.; Yin, Q.; Ruan, J.; Yu, X.; Egelman, E.; Wu, H. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015, 1, 15013. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.-W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Sauer, J.-D.; Witte, C.E.; Zemansky, J.; Hanson, B.; Lauer, P.; Portnoy, D.A. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010, 7, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Saiga, H.; Kitada, S.; Shimada, Y.; Kamiyama, N.; Okuyama, M.; Makino, M.; Yamamoto, M.; Takeda, K. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int. Immunol. 2012, 24, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenburg, S.; Brennan, G. Species-Specific Host–Virus Interactions: Implications for Viral Host Range and Virulence. Trends Microbiol. 2020, 28, 46–56. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Bruneau, R.C.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. https://doi.org/10.3390/biomedicines9070765
Yu H, Bruneau RC, Brennan G, Rothenburg S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines. 2021; 9(7):765. https://doi.org/10.3390/biomedicines9070765
Chicago/Turabian StyleYu, Huibin, Ryan C. Bruneau, Greg Brennan, and Stefan Rothenburg. 2021. "Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion" Biomedicines 9, no. 7: 765. https://doi.org/10.3390/biomedicines9070765
APA StyleYu, H., Bruneau, R. C., Brennan, G., & Rothenburg, S. (2021). Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines, 9(7), 765. https://doi.org/10.3390/biomedicines9070765