
Extra- and Intra-Cellular Mechanisms of Hepatic Stellate Cell Activation


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Extracellular Factors of HSC Activation
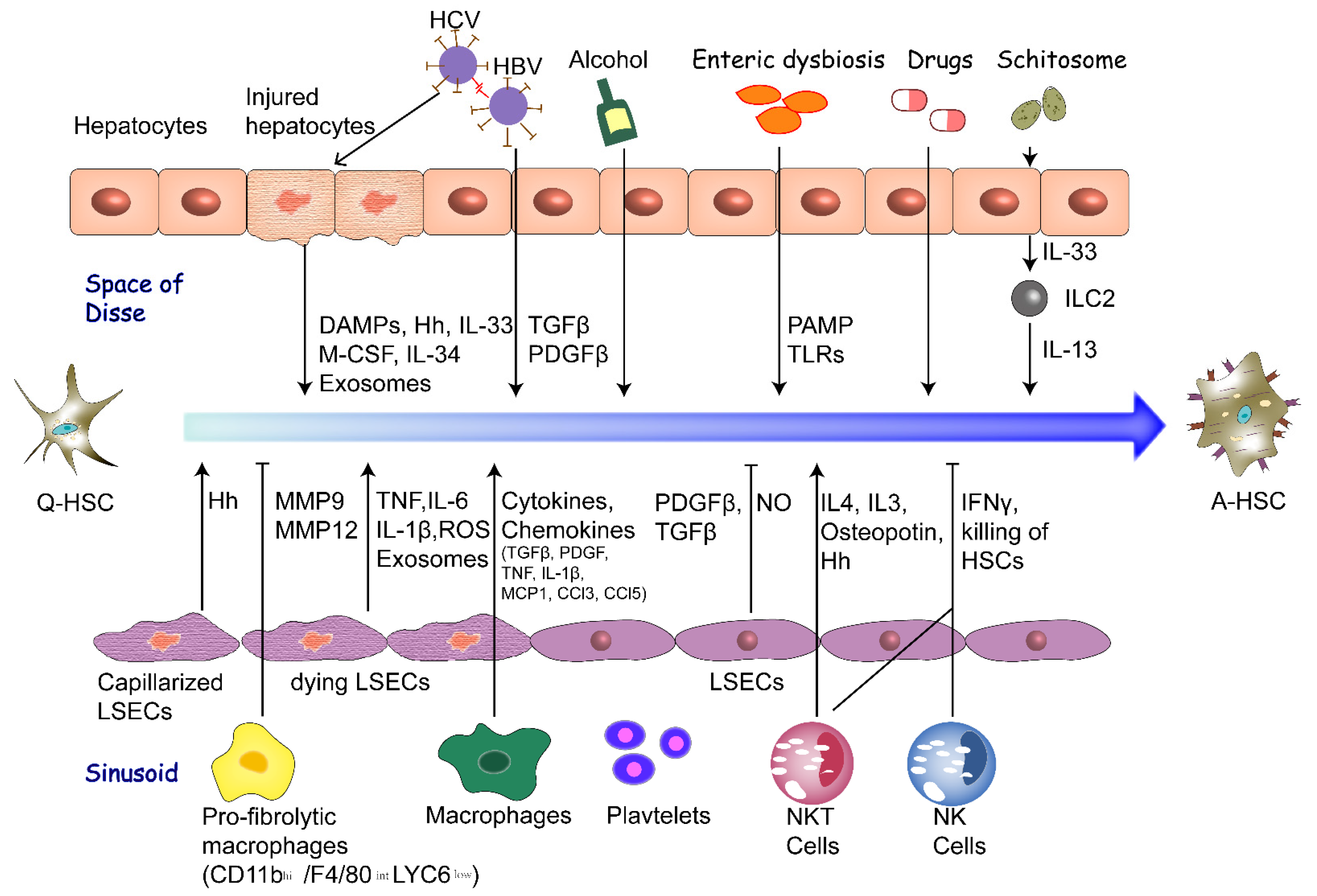
2.1. Hepatocytes
2.2. Liver Sinusoidal Endothelial Cells
2.3. Macrophages
2.4. Fibrogenic Cytokines
2.5. Altered ECM
2.6. Enteric Dysbiosis
2.7. Chronic Infection of Hepatitis Virus
2.8. Lipid Metabolism Disorder
2.9. Exosome and MicroRNA
2.10. Other Factors
3. Intracellular Signaling Pathways of HSC Activation
3.1. TGF-β/SMAD Pathway
3.2. Notch Signaling
3.3. Wnt/β-Catenin Signaling
3.4. Hedgehog Signaling
3.5. Hippo Signaling
3.6. Crosstalk of Intracellular Pathways
- Hippo and TGF-β/SMAD: It is demonstrated that YAP signaling works by promoting the binding of SMAD7 to activated TGF-β receptor type I, thereby eliminating downstream TGF-β signal transduction. At the same time, TAZ binds to SMAD2/3/4 heteromers in a TGF-β-dependent manner and recruits them into TGF-β response elements [121]. TAZ knockout experiments also show that TAZ plays a key role in the nuclear accumulation of SMAD2/3/4 complex in response to TGF-β and subsequent transactivation of target genes. In addition, The Hippo pathway scaffold protein RASSF1 is recruited by TGF-β to TGF-β receptor I, and is degraded by the E3 ubiquitin ligase ITCH co-recruited by the receptor, which in turn inactivates the MST/LATS kinase cascade and promotes YAP/SMAD2 interaction and subsequent nuclear translocation [121,122].
- TGF-β and Notch: Excessive activation of TGF-β regulates the Notch signaling pathway in the process of liver fibrosis in rats. Inhibiting the TGF-β signaling pathway can block the Notch signaling pathway, and Notch signaling can participate in the occurrence of liver fibrosis by activating the TGF-β/SMAD pathway. TGF-β inhibitor down-regulated the expression of Notch1, Hes1 and Hes5, and inhibited Notch signal mRNA and protein expression [123]. TGF-β1 also induced the high expression of Notch1, JAG1, Hes1 in HSC. The expression of the above-mentioned markers in mouse HSC was significantly reduced after TGF-β1 knockout. After blocking the Notch pathway with specific inhibitors, the expression of Notch1 and α-SMA in HSCs was significantly reduced. These results indicate that TGF-β1 signal controls the activation of HSCs by regulating the expression of Notch signaling pathway markers [124].
- Hedgehog and Hippo: The activation of the Hedgehog pathway promotes the post-transcriptional response of YAP by increasing the level of YAP protein, so Hedgehog signaling positively regulates YAP [125]. Blocking Hedgehog signaling can inhibit YAP activation in cultured HSCs, and downregulating YAP can inhibit YAP and Hedgehog-induced target gene expression, and inhibit HSC transdifferentiation into myofibroblasts, showing that the Hedgehog pathway can regulate the YAP protein of the regenerated liver in mice [126]. Previous studies have found that the Hedgehog pathway controls the HSCs activation by regulating cellular glycolysis. Conditional interruption of Hh signaling in myofibroblasts reduces the number of glycolytic myofibroblasts and the degree of liver fibrosis in mice [127]. Nevertheless, new research shows that the Hedgehog-YAP signaling pathway can promote the activation of HSCs by regulating the metabolism (i.e., the breakdown of glutamine) during the HSCs transdifferentiation into myofibroblasts [120]. Therefore, glutamine decomposition can control the accumulation of myofibroblasts in mice and may become a therapeutic target for liver fibrosis.
- Hedgehog and Notch: Activating the Notch pathway in HSCs can stimulate them to become myofibroblasts through a mechanism involving epithelial-mesenchymal transition, which needs to cross the typical Hedgehog pathway. It is suggested that when HSCs are converted to myofibroblasts, it activates Hh signal, undergoes epithelial to mesenchymal transition, and increases the expression of Notch signal. However, blocking Notch signaling in myofibroblasts can inhibit Hh signaling activity and cause mesenchymal epithelial transition; inhibiting Hh pathway can inhibit Notch signaling transduction and also induce mesenchymal epithelial transition [128].
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torres-Hernandez, A.; Wang, W.; Nikiforov, Y.; Tejada, K.; Torres, L.; Kalabin, A.; Wu, Y.; Haq, M.I.U.; Khan, M.Y.; Zhao, Z.; et al. Targeting SYK signaling in myeloid cells protects against liver fibrosis and hepatocarcinogenesis. Oncogene 2019, 38, 4512–4526. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; De Stafano, V.; Guo, X. Treatment of Patients with Cirrhosis. N. Engl. J. Med. 2016, 375, 2103–2104. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Z.; Zhang, Y.; Li, W.; Zheng, W.; Yu, J.; Wang, B.; Chen, L.; Zhuo, Q.; Chen, L.; et al. MicroRNA-212 activates hepatic stellate cells and promotes liver fibrosis via targeting SMAD7. Biochem. Biophys. Res. Commun. 2018, 496, 176–183. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.R.; Tian, Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J. Hepatol. 2019, 11, 412–420. [Google Scholar] [CrossRef]
- Lepreux, S.; Desmouliere, A. Human liver myofibroblasts during development and diseases with a focus on portal (myo)fibroblasts. Front. Physiol. 2015, 6, 173. [Google Scholar] [CrossRef]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, S.Y.; Ko, E.; Lee, J.H.; Yi, H.S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 3710. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. Incomplete Differentiation of Engrafted Bone Marrow Endothelial Progenitor Cells Initiates Hepatic Fibrosis in the Rat. Hepatology 2019, 69, 1259–1272. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927.e916. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kuo, L.M.; Wu, Y.H.; Chang, Y.C.; Lai, K.H.; Hwang, T.L. BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFbeta1-Activated Hepatic Stellate Cells. Biomedicines 2020, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, L.; Wei, R.; Ye, T.; Zhou, J.; Wen, M.; Men, R.; Aqeilan, R.I.; Peng, Y.; Yang, L. Twist1-induced miR-199a-3p promotes liver fibrosis by suppressing caveolin-2 and activating TGF-beta pathway. Signal Transduct. Target. Ther. 2020, 5, 75. [Google Scholar] [CrossRef]
- An, S.Y.; Petrescu, A.D.; DeMorrow, S. Targeting Certain Interleukins as Novel Treatment Options for Liver Fibrosis. Front. Pharmacol. 2021, 12, 645703. [Google Scholar] [CrossRef]
- Liu, Y.; Munker, S.; Mullenbach, R.; Weng, H.L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Meyer, C.; Muller, A.; Herweck, F.; Li, Q.; Mullenbach, R.; Mertens, P.R.; Dooley, S.; Weng, H.L. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J. Immunol. 2011, 187, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Sheng, L.; Chen, Z.; Jiang, L.; Su, H.; Yin, L.; Omary, M.B.; Rui, L. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology 2014, 60, 2065–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Kurokawa, T.; Zheng, Y.W.; Ohkohchi, N. Novel functions of platelets in the liver. J. Gastroenterol. Hepatol. 2016, 31, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Fan, W.; Liu, T.; Chen, W.; Hammad, S.; Longerich, T.; Hausser, I.; Fu, Y.; Li, N.; He, Y.; Liu, C.; et al. ECM1 Prevents Activation of Transforming Growth Factor beta, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology 2019, 157, 1352–1367.e1313. [Google Scholar] [CrossRef] [Green Version]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; You, Y.; Hua, M.; Wu, P.; Liu, Y.; Chen, Z.; Zhang, L.; Wei, H.; Li, Y.; Luo, M.; et al. Chlorophyllin Modulates Gut Microbiota and Inhibits Intestinal Inflammation to Ameliorate Hepatic Fibrosis in Mice. Front. Physiol. 2018, 9, 1671. [Google Scholar] [CrossRef] [Green Version]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C.; et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Zan, Y.; Zhang, Y.; Tien, P. Hepatitis B virus e antigen induces activation of rat hepatic stellate cells. Biochem. Biophys. Res. Commun. 2013, 435, 391–396. [Google Scholar] [CrossRef]
- Bai, Q.; An, J.; Wu, X.; You, H.; Ma, H.; Liu, T.; Gao, N.; Jia, J. HBV promotes the proliferation of hepatic stellate cells via the PDGF-B/PDGFR-beta signaling pathway in vitro. Int. J. Mol. Med. 2012, 30, 1443–1450. [Google Scholar] [CrossRef] [Green Version]
- Florimond, A.; Chouteau, P.; Bruscella, P.; Le Seyec, J.; Merour, E.; Ahnou, N.; Mallat, A.; Lotersztajn, S.; Pawlotsky, J.M. Human hepatic stellate cells are not permissive for hepatitis C virus entry and replication. Gut 2015, 64, 957–965. [Google Scholar] [CrossRef]
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Fremaux, I.; Croue, A.; Fouchard, I.; et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014, 60, 1879–1890. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.S.; Lee, Y.S.; Byun, J.S.; Seo, W.; Jeong, J.M.; Park, O.; Duester, G.; Haseba, T.; Kim, S.C.; Park, K.G.; et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology 2014, 60, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- O’Mahony, F.; Wroblewski, K.; O’Byrne, S.M.; Jiang, H.; Clerkin, K.; Benhammou, J.; Blaner, W.S.; Beaven, S.W. Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology 2015, 62, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Ye, Q.; Zhou, Y.; Zhao, C.; Xu, L.; Ping, J. Salidroside Inhibits CCl4-Induced Liver Fibrosis in Mice by Reducing Activation and Migration of HSC Induced by Liver Sinusoidal Endothelial Cell-Derived Exosomal SphK1. Front. Pharmacol. 2021, 12, 677810. [Google Scholar] [CrossRef]
- Ezhilarasan, D. MicroRNA interplay between hepatic stellate cell quiescence and activation. Eur. J. Pharmacol. 2020, 885, 173507. [Google Scholar] [CrossRef]
- Ma, L.; Yang, X.; Wei, R.; Ye, T.; Zhou, J.K.; Wen, M.; Men, R.; Li, P.; Dong, B.; Liu, L.; et al. MicroRNA-214 promotes hepatic stellate cell activation and liver fibrosis by suppressing Sufu expression. Cell Death Dis. 2018, 9, 718. [Google Scholar] [CrossRef]
- You, K.; Li, S.Y.; Gong, J.; Fang, J.H.; Zhang, C.; Zhang, M.; Yuan, Y.; Yang, J.; Zhuang, S.M. MicroRNA-125b Promotes Hepatic Stellate Cell Activation and Liver Fibrosis by Activating RhoA Signaling. Mol. Ther. Nucleic Acids 2018, 12, 57–66. [Google Scholar] [CrossRef]
- Song, L.Y.; Ma, Y.T.; Wu, C.F.; Wang, C.J.; Fang, W.J.; Liu, S.K. MicroRNA-195 Activates Hepatic Stellate Cells In Vitro by Targeting Smad7. Biomed. Res. Int. 2017, 2017, 1945631. [Google Scholar] [CrossRef] [Green Version]
- Page, A.; Paoli, P.P.; Hill, S.J.; Howarth, R.; Wu, R.; Kweon, S.M.; French, J.; White, S.; Tsukamoto, H.; Mann, D.A.; et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J. Hepatol. 2015, 62, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, C.; Tiniakos, D. Fibrosis and alcohol-related liver disease. J. Hepatol. 2019, 70, 294–304. [Google Scholar] [CrossRef]
- Ezhilarasan, D. Hepatotoxic potentials of methotrexate: Understanding the possible toxicological molecular mechanisms. Toxicology 2021, 458, 152840. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Oxidant Stress and Acetaminophen Hepatotoxicity: Mechanism-Based Drug Development. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef]
- Mannaerts, I.; Eysackers, N.; Anne van Os, E.; Verhulst, S.; Roosens, T.; Smout, A.; Hierlemann, A.; Frey, O.; Leite, S.B.; van Grunsven, L.A. The fibrotic response of primary liver spheroids recapitulates in vivo hepatic stellate cell activation. Biomaterials 2020, 261, 120335. [Google Scholar] [CrossRef]
- Chang, D.; Ramalho, L.N.; Ramalho, F.S.; Martinelli, A.L.; Zucoloto, S. Hepatic stellate cells in human schistosomiasis mansoni: A comparative immunohistochemical study with liver cirrhosis. Acta Trop. 2006, 97, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.P.; Ramm, G.A.; Robinson, M.W.; McManus, D.P.; Gobert, G.N. Schistosome-Induced Fibrotic Disease: The Role of Hepatic Stellate Cells. Trends Parasitol. 2018, 34, 524–540. [Google Scholar] [CrossRef]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Carthy, J.M. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef]
- Li, C.; Kong, Y.; Wang, H.; Wang, S.; Yu, H.; Liu, X.; Yang, L.; Jiang, X.; Li, L.; Li, L. Homing of bone marrow mesenchymal stem cells mediated by sphingosine 1-phosphate contributes to liver fibrosis. J. Hepatol. 2009, 50, 1174–1183. [Google Scholar] [CrossRef]
- Lei, X.F.; Fu, W.; Kim-Kaneyama, J.R.; Omoto, T.; Miyazaki, T.; Li, B.; Miyazaki, A. Hic-5 deficiency attenuates the activation of hepatic stellate cells and liver fibrosis through upregulation of Smad7 in mice. J. Hepatol. 2016, 64, 110–117. [Google Scholar] [CrossRef]
- Yang, Y.M.; Noureddin, M.; Liu, C.; Ohashi, K.; Kim, S.Y.; Ramnath, D.; Powell, E.E.; Sweet, M.J.; Roh, Y.S.; Hsin, I.F.; et al. Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 2019, 11, eaat9284. [Google Scholar] [CrossRef]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.; Cummings, R.D.; Drickamer, K.; Feizi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.; et al. Galectins: A family of animal beta-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Akahani, S.; Nangia-Makker, P.; Inohara, H.; Kim, H.R.; Raz, A. Galectin-3: A novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 1997, 57, 5272–5276. [Google Scholar]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.H.; Chen, Y.L.; Lee, K.H.; Chang, C.C.; Cheng, T.M.; Wu, S.Y.; Tu, C.C.; Tsui, W.L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 11006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Zhao, Y.X.; Wei, D.; Li, Y.L.; Zhang, Y.; Wu, J.; Xu, J.; Chen, C.; Tang, H.; Zhang, W.; et al. HAb18G/CD147 promotes activation of hepatic stellate cells and is a target for antibody therapy of liver fibrosis. J. Hepatol. 2012, 57, 1283–1291. [Google Scholar] [CrossRef]
- Kinoshita, K.; Iimuro, Y.; Otogawa, K.; Saika, S.; Inagaki, Y.; Nakajima, Y.; Kawada, N.; Fujimoto, J.; Friedman, S.L.; Ikeda, K. Adenovirus-mediated expression of BMP-7 suppresses the development of liver fibrosis in rats. Gut 2007, 56, 706–714. [Google Scholar] [CrossRef]
- Hao, Z.M.; Cai, M.; Lv, Y.F.; Huang, Y.H.; Li, H.H. Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Mol. Ther. 2012, 20, 2043–2051. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.L.; Zuo, S.; Lu, S.; Hu, R.H.; Lu, Y.Y.; Yang, J.; Deng, K.S.; Wu, Y.T.; Mu, M.; Zhu, J.J.; et al. Bone morphogenetic protein-7 represses hepatic stellate cell activation and liver fibrosis via regulation of TGF-β/Smad signaling pathway. World J. Gastroenterol. 2019, 25, 4222–4234. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Wacker, E.; Dorn, C.; Koch, A.; Saugspier, M.; Thasler, W.E.; Hartmann, A.; Bosserhoff, A.K.; Hellerbrand, C. Enhanced expression of BMP6 inhibits hepatic fibrosis in non-alcoholic fatty liver disease. Gut 2015, 64, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Geisler, F.; Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 2015, 61, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Sawitza, I.; Kordes, C.; Reister, S.; Haussinger, D. The niche of stellate cells within rat liver. Hepatology 2009, 50, 1617–1624. [Google Scholar] [CrossRef]
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef]
- Zhu, C.; Ho, Y.J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.S.; Lowe, S.W.; Pajvani, U.B. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J. Hepatol. 2021, 74, 613–626. [Google Scholar] [CrossRef]
- Richter, L.R.; Wan, Q.; Wen, D.; Zhang, Y.; Yu, J.; Kang, J.K.; Zhu, C.; McKinnon, E.L.; Gu, Z.; Qiang, L.; et al. Targeted Delivery of Notch Inhibitor Attenuates Obesity-Induced Glucose Intolerance and Liver Fibrosis. ACS Nano 2020, 14, 6878–6886. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [Green Version]
- Barrott, J.J.; Cash, G.M.; Smith, A.P.; Barrow, J.R.; Murtaugh, L.C. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 12752–12757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Shackel, N.A.; McGuinness, P.H.; Abbott, C.A.; Gorrell, M.D.; McCaughan, G.W. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut 2001, 49, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Parsons, C.J.; Stefanovic, B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 2006, 45, 401–409. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, Tolerability, and Preliminary Efficacy of the Anti-Fibrotic Small Molecule PRI-724, a CBP/β-Catenin Inhibitor, in Patients with Hepatitis C Virus-related Cirrhosis: A Single-Center, Open-Label, Dose Escalation Phase 1 Trial. EBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Zhu, C.; Tabas, I.; Schwabe, R.F.; Pajvani, U.B. Maladaptive regeneration—The reawakening of developmental pathways in NASH and fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 131–142. [Google Scholar] [CrossRef]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Hedgehog signalling in liver pathophysiology. J. Hepatol. 2018, 68, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Sicklick, J.K.; Li, Y.X.; Melhem, A.; Schmelzer, E.; Zdanowicz, M.; Huang, J.; Caballero, M.; Fair, J.H.; Ludlow, J.W.; McClelland, R.E.; et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G859–G870. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Kruger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Investig. 2013, 123, 2380–2394. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [Green Version]
- Swiderska-Syn, M.; Syn, W.K.; Xie, G.; Krüger, L.; Machado, M.V.; Karaca, G.; Michelotti, G.A.; Choi, S.S.; Premont, R.T.; Diehl, A.M. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut 2014, 63, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.D.; Suzuki, A.; Abdelmalek, M.F.; Burchette, J.L.; Diehl, A.M. Treatment response in the PIVENS trial is associated with decreased Hedgehog pathway activity. Hepatology 2015, 61, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator with PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2020, 71, 1813–1830. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.X.; Yao, Y.; Bu, F.T.; Chen, Y.; Wu, Y.T.; Yang, Y.; Chen, X.; Zhu, Y.; Wang, Q.; Pan, X.Y.; et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2019, 107, 29–40. [Google Scholar] [CrossRef]
- Anakk, S.; Bhosale, M.; Schmidt, V.A.; Johnson, R.L.; Finegold, M.J.; Moore, D.D. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013, 5, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Einwallner, E.; Claudel, T.; Trauner, M. PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells. Int. J. Mol. Sci. 2020, 21, 8711. [Google Scholar] [CrossRef]
- Konishi, T.; Schuster, R.M.; Lentsch, A.B. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G471–G482. [Google Scholar] [CrossRef]
- Driskill, J.H.; Pan, D. The Hippo Pathway in Liver Homeostasis and Pathophysiology. Annu. Rev. Pathol. 2021, 16, 299–322. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Harvey, L.D.; Chan, S.Y. YAPping About Glutaminolysis in Hepatic Fibrosis. Gastroenterology 2018, 154, 1231–1233. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e1413. [Google Scholar] [CrossRef] [Green Version]
- Pefani, D.E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S.; O’Neill, E. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Hu, J.; Pan, J.; Wang, Y.; Hu, G.; Zhou, J.; Mei, L.; Xiong, W.C. YAP stabilizes SMAD1 and promotes BMP2-induced neocortical astrocytic differentiation. Development 2016, 143, 2398–2409. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shen, R.W.; Han, B.; Li, Z.; Xiong, L.; Zhang, F.Y.; Cong, B.B.; Zhang, B. Notch signaling mediated by TGF-β/Smad pathway in concanavalin A-induced liver fibrosis in rats. World J. Gastroenterol. 2017, 23, 2330–2336. [Google Scholar] [CrossRef]
- Aimaiti, Y.; Yusufukadier, M.; Li, W.; Tuerhongjiang, T.; Shadike, A.; Meiheriayi, A.; Abudusalamu, A.; Wang, H.; Tuerganaili, A.; Shao, Y.; et al. TGF-β1 signaling activates hepatic stellate cells through Notch pathway. Cytotechnology 2019, 71, 881–891. [Google Scholar] [CrossRef]
- Tariki, M.; Dhanyamraju, P.K.; Fendrich, V.; Borggrefe, T.; Feldmann, G.; Lauth, M. The Yes-associated protein controls the cell density regulation of Hedgehog signaling. Oncogenesis 2014, 3, e112. [Google Scholar] [CrossRef] [Green Version]
- Swiderska-Syn, M.; Xie, G.; Michelotti, G.A.; Jewell, M.L.; Premont, R.T.; Syn, W.K.; Diehl, A.M. Hedgehog regulates yes-associated protein 1 in regenerating mouse liver. Hepatology 2016, 64, 232–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e1311. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Krüger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Zeng, J.; Xing, L.; Li, C. Extra- and Intra-Cellular Mechanisms of Hepatic Stellate Cell Activation. Biomedicines 2021, 9, 1014. https://doi.org/10.3390/biomedicines9081014
Yan Y, Zeng J, Xing L, Li C. Extra- and Intra-Cellular Mechanisms of Hepatic Stellate Cell Activation. Biomedicines. 2021; 9(8):1014. https://doi.org/10.3390/biomedicines9081014
Chicago/Turabian StyleYan, Yufei, Jiefei Zeng, Linhao Xing, and Changyong Li. 2021. "Extra- and Intra-Cellular Mechanisms of Hepatic Stellate Cell Activation" Biomedicines 9, no. 8: 1014. https://doi.org/10.3390/biomedicines9081014
APA StyleYan, Y., Zeng, J., Xing, L., & Li, C. (2021). Extra- and Intra-Cellular Mechanisms of Hepatic Stellate Cell Activation. Biomedicines, 9(8), 1014. https://doi.org/10.3390/biomedicines9081014