
GRP78 Overexpression Triggers PINK1-IP3R-Mediated Neuroprotective Mitophagy

 ,
,  , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Surgical Procedures
2.2. Construction, Purification, and Infection with Viral Vectors
2.3. Sample Preparation and Proteomic Analysis
2.4. Immunohistochemistry and Image Analysis
2.5. Western Blot
2.6. In Vitro Model
2.7. Nucleofection
2.8. Measurement of Mitochondrial Superoxide Production and ΔψM
2.9. Electrochemical Measurement of Oxygen Consumption
2.10. Mitochondrial and Cytosolic Fractionation
2.11. Transmission Electron Microscopy
2.12. Bioinformatics and Statistics
3. Results
3.1. Text
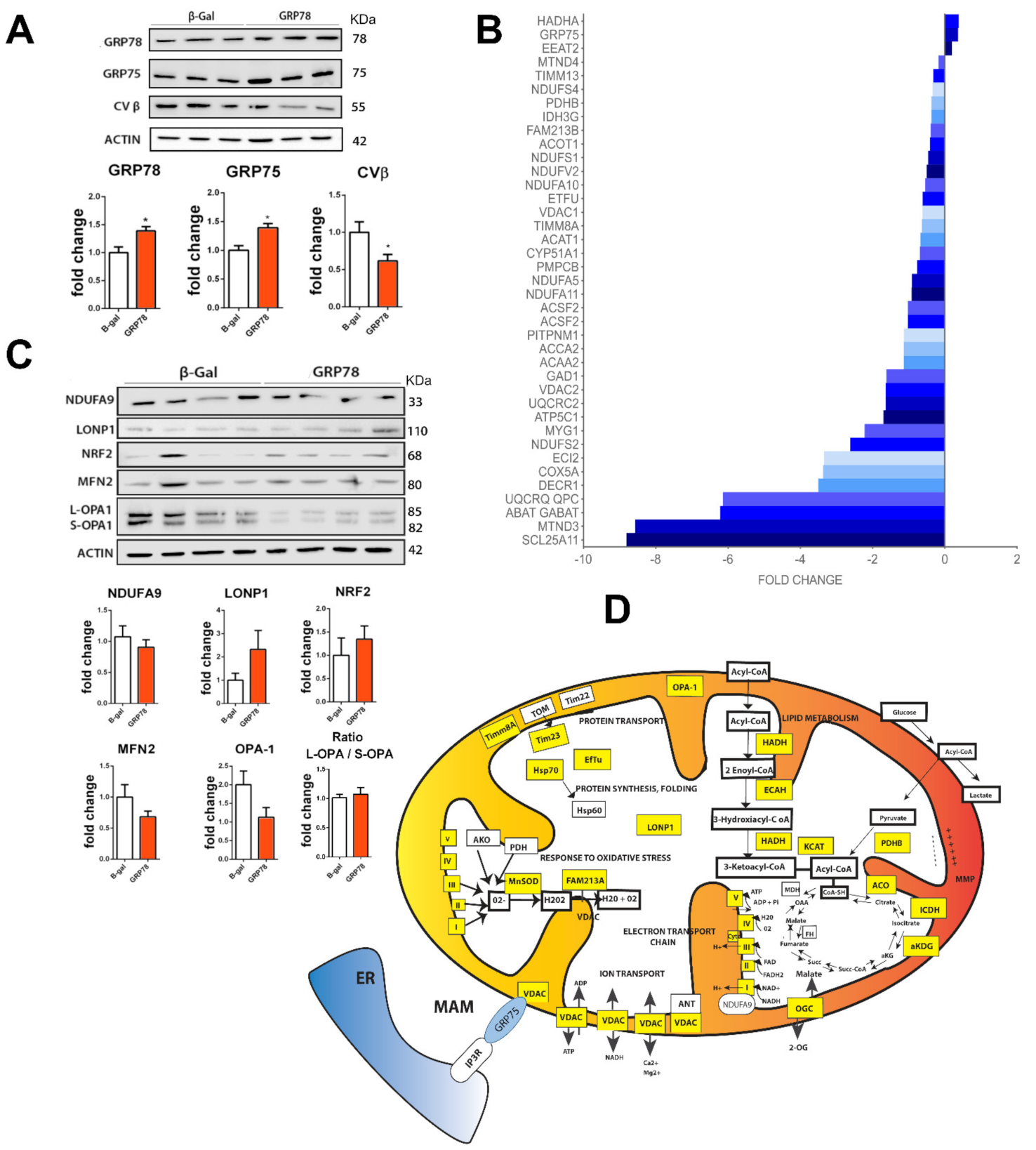
3.1.1. Proteomic Analysis of GRP78 Overexpression Revealed Mitochondria as the Main Target
3.1.2. GRP78 Overexpression Restores Damaged Mitochondrial Function
3.1.3. Mitophagy Induction by GRP78 Overexpression Mediates Neuroprotection
3.1.4. Neuroprotection Mediated by GRP78 Depends on PINK1 and IP3R
4. Discussion
Study Limitations and Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Special Statement
References
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Forés, J.; Herrando-Grabulosa, M.; Valls, R.; Leiva-Rodríguez, T.; Galea, E.; González-Pérez, F.; Navarro, X.; Petegnief, V.; Bosch, A.; et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci. Rep. 2018, 8, 1879. [Google Scholar] [CrossRef] [Green Version]
- Romeo-Guitart, D.; Forés, J.; Navarro, X.; Casas, C. Boosted Regeneration and Reduced Denervated Muscle Atrophy by NeuroHeal in a Pre-clinical Model of Lumbar Root Avulsion with Delayed Reimplantation. Sci. Rep. 2017, 7, 12028. [Google Scholar] [CrossRef] [Green Version]
- Casas, C. Grp78 at the Centre of the Stage in Neurodegeneration and Cancer. Front. Neurosci. 2017, 11, 1–15. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topkudoisomerase inhibitors. Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Printsev, I.; Curiel, D.; Carraway, K.L. Membrane Protein Quantity Control at the Endoplasmic Reticulum. J. Membr. Biol. 2016, 250, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.-W.; Sun, F.-C.; Cho, J.-H.; Lin, C.-C.; Liu, P.-F.; Chen, P.-Y.; Chang, M.D.-T.; Fu, H.-W.; Lai, Y.-K. GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum stress-induced apoptosis. J. Cell. Physiol. 2008, 215, 627–635. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, R. Heat shock 70 kDa protein 5/glucose-regulated protein 78 “AMP”ing up autophagy. Autophagy 2012, 8, 1827–1829. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y.; Newton, I.P.; Zhang, L.; Ji, P.; Li, Z. GRP78 is implicated in the modulation of tumor aerobic glycolysis by promoting autophagic degradation of IKKβ. Cell. Signal. 2015, 27, 1237–1245. [Google Scholar] [CrossRef]
- Cha-Molstad, H.; Yu, J.E.; Lee, S.H.; Kim, J.G.; Sung, K.S.; Hwang, J.; Yoo, Y.D.; Lee, Y.J.; Kim, S.T.; Lee, D.H.; et al. Modulation of SQSTM1/p62 activity by N-terminal arginylation of the endoplasmic reticulum chaperone HSPA5/GRP78/BiP. Autophagy 2016, 12, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Cha-Molstad, H.; Sung, K.S.; Hwang, J.; Kim, K.A.; Yu, J.E.; Yoo, Y.D.; Jang, J.M.; Han, D.H.; Molstad, M.; Kim, J.G.; et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 2015, 17, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Abdel Malek, M.A.Y.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; Abd El-Azeem, H.G.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Hong, S.-K.; Wu, P.-K.; Richards, A.L.; Jackson, W.T.; Park, J.-I. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp. Cell Res. 2014, 327, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Mimura, N.; Kashio, M.; Koseki, H.; Aoe, T. Late-onset of spinal neurodegeneration in knock-in mice expressing a mutant BIP. PLoS ONE 2014, 9, e112837. [Google Scholar] [CrossRef]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Oida, Y.; Hamanaka, J.; Hyakkoku, K.; Shimazawa, M.; Kudo, T.; Imaizumi, K.; Yasuda, T.; Hara, H. Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci. Lett. 2010, 484, 43–46. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signaling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.B.; Xu, L.J.; Emery, J.F.; Lee, A.S.; Giffard, R.G. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion 2011, 11, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penas, C.; Font-Nieves, M.; Forés, J.; Petegnief, V.; Planas, A.; Navarro, X.; Casas, C. Autophagy, and BiP level decrease are early key events in retrograde degeneration of motoneurons. Cell Death Differ. 2011, 18, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Penas, C.; Pascual-Font, A.; Mancuso, R.; Forés, J.; Casas, C.; Navarro, X. Sigma receptor agonist 2-(4-morpholinethyl)1 phenylcyclohexanecarboxylate (Pre084) increases GDNF and BiP expression and promotes neuroprotection after root avulsion injury. J. Neurotrauma 2011, 28, 831–840. [Google Scholar] [CrossRef]
- Guzmán-Lenis, M.-S.; Navarro, X.; Casas, C. Selective sigma receptor agonist 2-(4-morpholinethyl)1-phenylcyclohexanecarboxylate (PRE084) promotes neuroprotection and neurite elongation through protein kinase C (PKC) signaling on motoneurons. Neuroscience 2009, 162, 31–38. [Google Scholar] [CrossRef]
- Louessard, M.; Bardou, I.; Lemarchand, E.; Thiebaut, A.M.; Parcq, J.; Leprince, J.; Terrisse, A.; Carraro, V.; Fafournoux, P.; Bruhat, A.; et al. Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death Differ. 2017, 24, 1518–1529. [Google Scholar] [CrossRef] [Green Version]
- Paz Gavilán, M.; Vela, J.; Castaño, A.; Ramos, B.; del Río, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef]
- Casas, C.; Isus, L.; Herrando-Grabulosa, M.; Mancuso, F.M.; Borrás, E.; Sabidó, E.; Forés, J.; Aloy, P. Network-based proteomic approaches reveal the neurodegenerative, neuroprotective and pain-related mechanisms involved after retrograde axonal damage. Sci. Rep. 2015, 5, 9185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiva-Rodríguez, T.; Romeo-Guitart, D.; Marmolejo-Martínez-Artesero, S.; Herrando-Grabulosa, M.; Bosch, A.; Forés, J.; Casas, C. ATG5 overexpression is neuroprotective and attenuates cytoskeletal and vesicle-Trafficking alterations in axotomized motoneurons article. Cell Death Dis. 2018, 9, 626. [Google Scholar] [CrossRef] [PubMed]
- Penas, C.; Casas, C.; Robert, I.; Forés, J.; Navarro, X. Cytoskeletal and activity-related changes in spinal motoneurons after root avulsion. J. Neurotrauma 2009, 26, 763–779. [Google Scholar] [CrossRef]
- Marmolejo-Martínez-Artesero, S.; Casas, C.; Romeo-Guitart, D. Endogenous Mechanisms of Neuroprotection: To Boost or Not to Boost. Cells 2021, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Loeb, J.E.; Cordier, W.S.; Harris, M.E.; Weitzman, M.D.; Hope, T.J. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: Implications for gene therapy. Human Gene Ther. 1999, 10, 2295–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Piedra, J.; Ontiveros, M.; Miravet, S.; Penalva, C.; Monfar, M.; Chillon, M. Development of a rapid, robust, and universal picogreen-based method to titer adeno-associated vectors. Human Gene Ther. Methods 2015, 26, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Homs, J.; Pagès, G.; Ariza, L.; Casas, C.; Chillón, M.; Navarro, X.; Bosch, A. Intrathecal administration of IGF-I by AAVrh10 improves sensory and motor deficits in a mouse model of diabetic neuropathy. Mol. Ther. Methods Clin. Dev. 2014, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cariño, C.; Duffy, C.; Sánchez-Chardi, A.; McNeilly, F.; Allan, G.M.; Segalés, J. Porcine circovirus type 2 morphogenesis in a clone derived from the l35 lymphoblastoid cell line. J. Comp. Pathol. 2011, 144, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.; Chen, J.-L.; Wang, Q.; Shao, W.; Qi, B. Modulation of Mitochondrial Dynamics in Neurodegenerative Diseases: An Insight into Prion Diseases. Front. Aging Neurosci. 2018, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Wang, L.; Qi, H.; Tang, Y.; Shen, H.-M. Post-translational Modifications of Key Machinery in the Control of Mitophagy. Trends Biochem. Sci. 2020, 45, 58–75. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Wang, L.; Cho, Y.-L.; Tang, Y.; Wang, J.; Park, J.-E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo-Guitart, D.; Leiva-Rodriguez, T.; Espinosa-Alcantud, M.; Sima, N.; Vaquero, A.; Dominguez-Martin, H.; Ruano, D.; Casas, C. SIRT1 activation with neuroheal is neuroprotective but SIRT2 inhibition with AK7 is detrimental for disconnected motoneurons. Cell Death Dis. 2018, 9, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funes, H.A.; Apostolova, N.; Alegre, F.; Blas-Garcia, A.; Alvarez, A.; Marti-Cabrera, M.; Esplugues, J.V. Neuronal Bioenergetics and Acute Mitochondrial Dysfunction: A Clue to Understanding the Central Nervous System Side Effects of Efavirenz. J. Infect. Dis. 2014, 210, 1385–1395. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.-E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 1997, 174, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Sampieri, A.; Santoyo, K.; Asanov, A.; Vaca, L. Association of the IP3R to STIM1 provides a reduced intraluminal calcium microenvironment, resulting in enhanced store-operated calcium entry. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Alexandre, S.; Cao, X.; Lee, A.S. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J. Biol. Chem. 1993, 268, 12003–12009. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Casas, C. Network-centric medicine for peripheral nerve injury: Treating the whole to boost endogenous mechanisms of neuroprotection and regeneration. Neural Regen. Res. 2019, 14, 1122–1128. [Google Scholar]
- Romeo-Guitart, D.; Marcos-DeJuana, C.; Marmolejo-Martínez-Artesero, S.; Navarro, X.; Casas, C. Novel neuroprotective therapy with NeuroHeal by autophagy induction for damaged neonatal motoneurons. Theranostics 2020, 10, 5154–5168. [Google Scholar] [CrossRef]
- Henderson, B.; Martin, A.C.R. Protein moonlighting: A new factor in biology and medicine. Biochem. Soc. Trans. 2014, 42, 1671–1678. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.-J.; Piao, Y.; Pak, Y.K. Endoplasmic reticulum stress impairs insulin signaling through mitochondrial damage in SH-SY5Y cells. Neurosignals 2012, 20, 265–280. [Google Scholar] [CrossRef]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [Green Version]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miharada, K.; Karlsson, G.; Rehn, M.; Rörby, E.; Siva, K.; Cammenga, J.; Karlsson, S. Cripto Regulates Hematopoietic Stem Cells as a Hypoxic-Niche-Related Factor through Cell Surface Receptor GRP78. Cell Stem Cell 2011, 9, 330–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-Differentiation Confers Multidrug Resistance Via Noncanonical PERK-Nrf2 Signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.W.; Chen, Y.S.; Tsay, Y.G.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Hung, K.F.; Lin, C.H.; Huang, T.Y.; Kao, S.Y.; et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 4, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, X.; Lu, Y.; Duan, C.; Gao, G.; Lu, L.; Yang, H. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017, 8, e3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Hemachandra Reddy, P. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef] [PubMed]
- Gaki, G.S.; Papavassiliou, A.G. Oxidative Stress-Induced Signaling Pathways Implicated in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2014, 16, 217–230. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2017, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Macvicar, T.D.B.; Mannack, L.V.J.C.; Lees, R.M.; Lane, J.D. Targeted siRNA screens identify ER-to-mitochondrial calcium exchange in autophagy and mitophagy responses in RPE1 cells. Int. J. Mol. Sci. 2015, 16, 13356–13380. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.-C.; Wei, S.; Li, C.-W.; Chang, Y.-S.; Chao, C.-C.; Lai, Y.-K. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem. J. 2006, 396, 31–39. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leiva-Rodríguez, T.; Romeo-Guitart, D.; Herrando-Grabulosa, M.; Muñoz-Guardiola, P.; Polo, M.; Bañuls, C.; Petegnief, V.; Bosch, A.; Lizcano, J.M.; Apostolova, N.; et al. GRP78 Overexpression Triggers PINK1-IP3R-Mediated Neuroprotective Mitophagy. Biomedicines 2021, 9, 1039. https://doi.org/10.3390/biomedicines9081039
Leiva-Rodríguez T, Romeo-Guitart D, Herrando-Grabulosa M, Muñoz-Guardiola P, Polo M, Bañuls C, Petegnief V, Bosch A, Lizcano JM, Apostolova N, et al. GRP78 Overexpression Triggers PINK1-IP3R-Mediated Neuroprotective Mitophagy. Biomedicines. 2021; 9(8):1039. https://doi.org/10.3390/biomedicines9081039
Chicago/Turabian StyleLeiva-Rodríguez, Tatiana, David Romeo-Guitart, Mireia Herrando-Grabulosa, Pau Muñoz-Guardiola, Miriam Polo, Celia Bañuls, Valerie Petegnief, Assumpció Bosch, Jose Miguel Lizcano, Nadezda Apostolova, and et al. 2021. "GRP78 Overexpression Triggers PINK1-IP3R-Mediated Neuroprotective Mitophagy" Biomedicines 9, no. 8: 1039. https://doi.org/10.3390/biomedicines9081039
APA StyleLeiva-Rodríguez, T., Romeo-Guitart, D., Herrando-Grabulosa, M., Muñoz-Guardiola, P., Polo, M., Bañuls, C., Petegnief, V., Bosch, A., Lizcano, J. M., Apostolova, N., Forés, J., & Casas, C. (2021). GRP78 Overexpression Triggers PINK1-IP3R-Mediated Neuroprotective Mitophagy. Biomedicines, 9(8), 1039. https://doi.org/10.3390/biomedicines9081039