
Poloxamer 188 Exerts Direct Protective Effects on Mouse Brain Microvascular Endothelial Cells in an In Vitro Traumatic Brain Injury Model



Abstract
1. Introduction
- (1)
- Develop a suitable in vitro compression-type TBI model using brain endothelial cells to test for potential treatments.
- (2)
- Evaluate direct effects of P188 given upon reoxygenation in an in vitro compression-type TBI model, applying mild-to-moderate and severe injury.
- (3)
- Assess protective effects of P188 post-treatment after excessive damage in vitro.
- (4)
- Test for osmotic effects by using polyethylene glycol (PEG) with a similar molecular weight as P188 serving as control.
2. Materials and Methods
2.1. Cell Culture
2.2. Compression-Type TBI Model
2.3. Cell Number/Viability
2.4. Cytotoxicity/Membrane Damage via Lactate Dehydrogenase Release
2.5. Metabolic Activity
2.6. Total NO Production
2.7. Statistics
3. Results
3.1. Effect of P188 after Mild-to-Moderate Injury
3.1.1. Cell Number/Viability
3.1.2. Cytotoxicity/Membrane Damage via LDH Release
3.1.3. Metabolic Activity
3.1.4. Total NO Production
3.2. Effect of P188 after Severe Injury
3.2.1. Cell Number/Viability
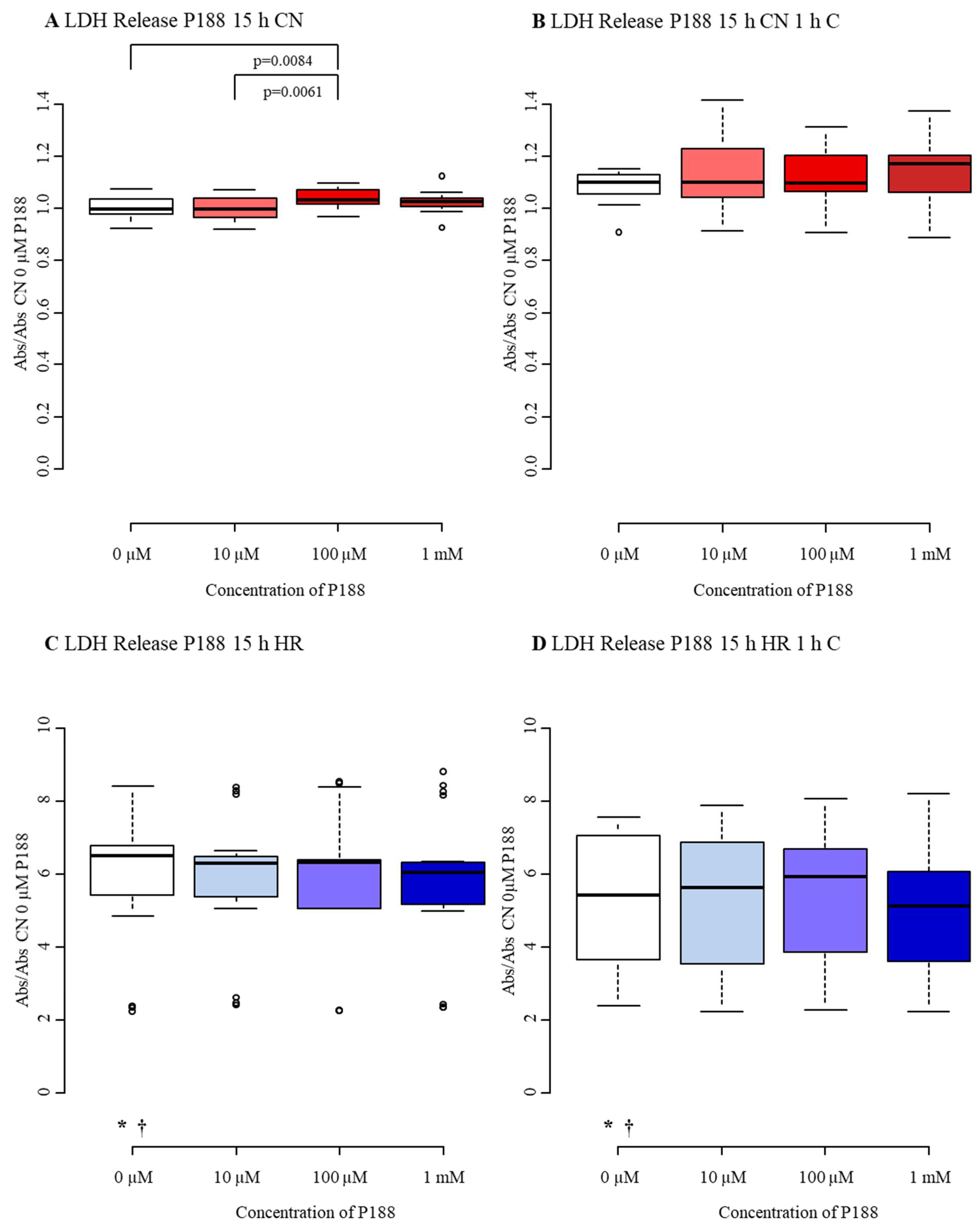
3.2.2. Cytotoxicity/Membrane Damage via LDH Release
3.2.3. Metabolic Activity
3.3. Diminished Effect of PEG after Mild-to-Moderate Injury
3.3.1. Cell Number/Viability
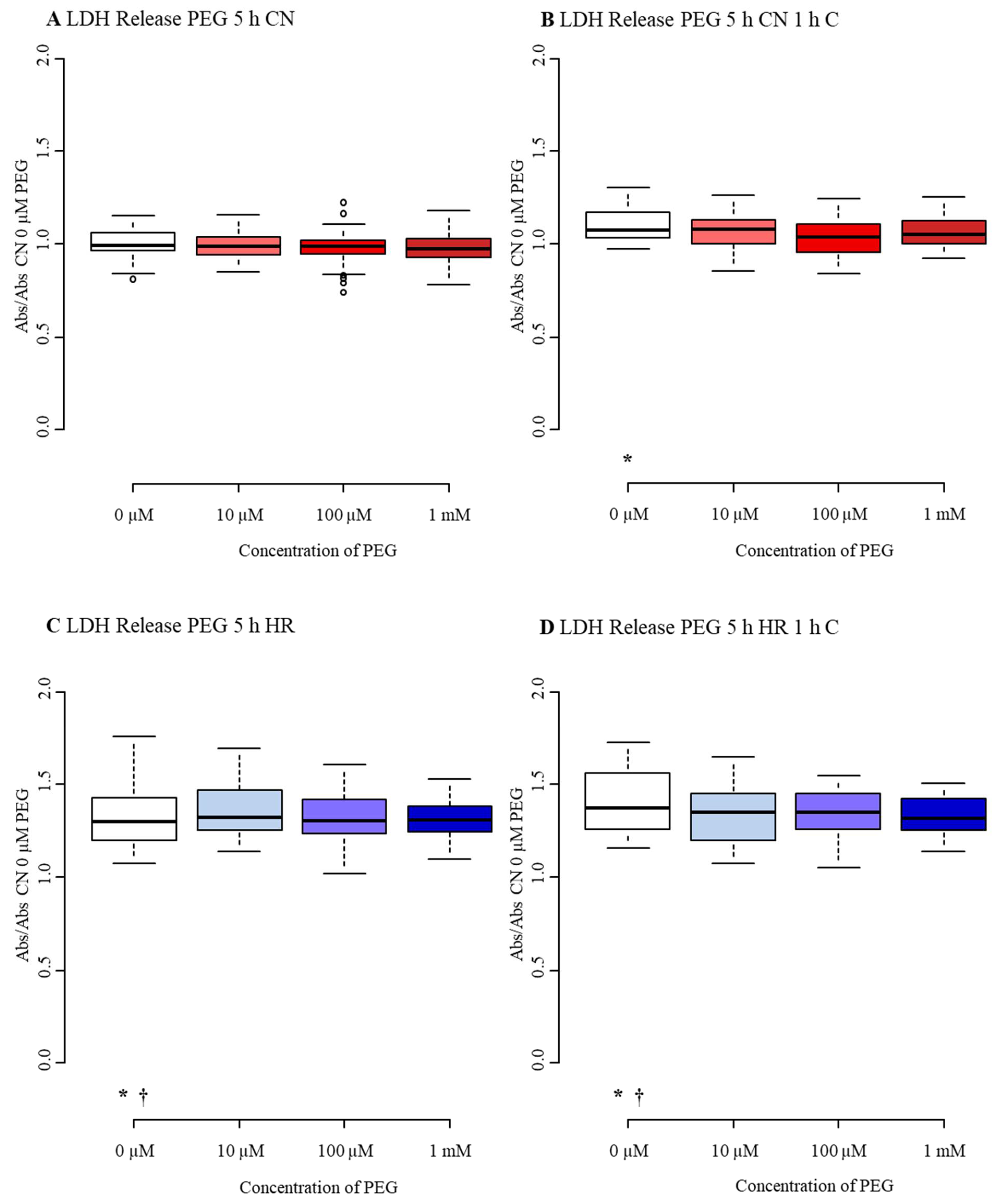
3.3.2. Cytotoxicity/Membrane Damage via LDH Release
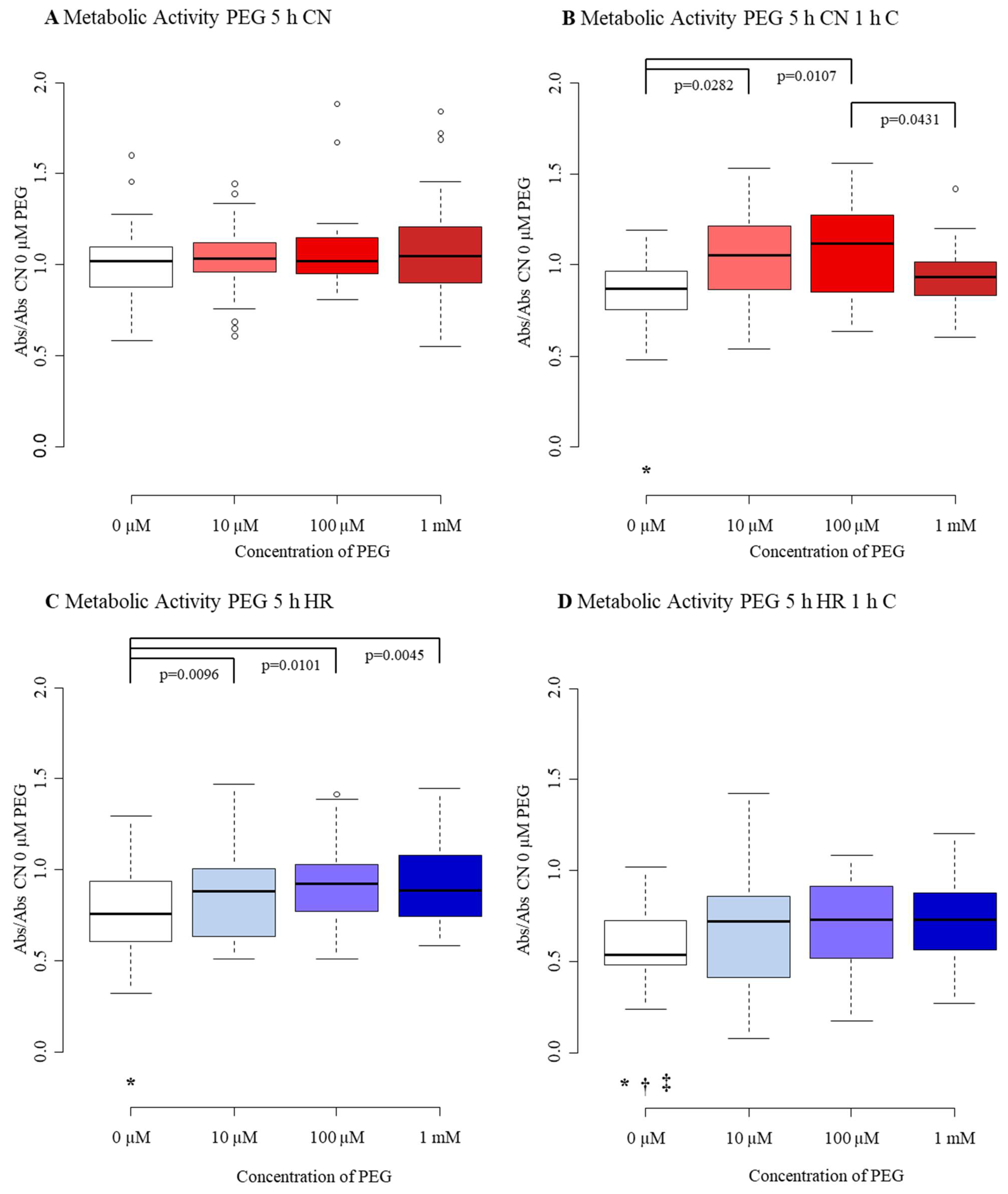
3.3.3. Metabolic Activity
4. Discussion
4.1. Cell Number/Viability
4.2. Cytotoxicity/Membrane Damage
4.3. Metabolic Activity
4.4. Total NO Production
4.5. Limitations
- We used an in vitro model which might not exactly mimic the complex pathogenesis of TBI in vivo. Morrison et al., though, suggested that in vitro models can predict effects in vivo in over 88% [69]. Even though 88% is quite high, some in vitro models do not adequately predict in vivo findings. However, in vitro models are reproducible, well-controlled, and can be performed within specific environments, without systemic confounders [69].
- MBEC were investigated. Even though mice are often used to mimic processes occurring in humans, they might not display the exact functioning of human brain microvascular endothelial cells. Song et al. found differences between mouse and human brain vasculature, that might be crucial for drug delivery and disease [70]. Interestingly, emerging approaches to address this limitation include an in vivo human blood vessel transplantation model [71].
- Although we were able to see significant effects of P188, some of these effects seem to be small in size, which might possibly reduce their relevance in preclinical studies. Nevertheless, we studied the effects on endothelial cells only, without taking into account that other cells of the neurovascular unit, neurons, and inflammatory cells may contribute to endothelial functioning and the effect of P188 in vivo. In addition, even small protective effects on endothelial cells might have amplified downstream effects on effector cells such as neurons as recently shown with cardiomyocytes [72].
- Furthermore, MBEC in the center of a well suffered more mechanical injury than cells growing at the margins of a well, as the rods of the compression devices were placed in the center. This might also have contributed to relatively high data variations observed in some sets of data.
- Another important limitation of this observational study is the absence of more detailed mechanistic experiments, which might further elucidate the underlying pathways of P188 protection in vitro in the future.
- Even though other hypoxia times might be suitable for MBEC [26], we only examined the effect of P188 on MBEC exposed to hypoxia for 5 and 15 h.
- Moreover, we did not observe the possible long-term effect of P188 administration. However, Gu et al. showed that daily intraperitoneal injection of P188 can enhance long-term outcomes after I/R injury [21]. In an in vitro blast-TBI model, endothelial cells were treated with P188 for 6 h with beneficial effects [19]. Consequently, the effect of P188 might differ when P188 is administered for a longer period than 2 h.
- Lastly, some of the protective effects of P188 have been described previously in various different models. However, the study by Inyang et al. [19] is the only one describing P188′s protective effect on isolated brain endothelial cells in the context of TBI. In contrast to our study that aimed to assess cellular functioning and cell survival after compression-injury in a hypoxia-reoxygenation model, though, they observed increased permeability, superoxide levels, and inflammatory trauma after blast-induced micro-cavitation, which included neither hypoxic nor reoxygenation injuries. Moreover, our study focused on very different endpoints, including but not limited to the effects of P188 on the production of NO which has not been described previously either and, thus, may address a possible mechanistic pathway for P188. Additionally, we are the first to show that the effect of P188 on MBEC when applied upon reoxygenation depends on the severity of the injury applied, with a decreasing effect after severe injury, limiting the protection by P188 to mild-to-moderate injury. Hence, our in vitro study does add several novel aspects to the current literature.
4.6. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Number/Viability 5 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 0.99 2,3 | 0.93 1,4 | 0.91 1,4 | 1.03 2,3 |
| (0.93, 1.06) | (0.86, 1.02) | (0.83, 0.99) | (0.91, 1.11) | |
| CN+C | 0.92 2,4,* | 0.53 1 | 0.65 | 0.47 1 |
| (0.39, 1.11) | (0.29, 0.77) | (0.30, 0.94) | (0.22, 0.74) | |
| HR-C | 0.86 4,* | 0.86 4 | 0.87 4 | 0.91 1,2,3 |
| (0.78, 0.97) | (0.76, 0.94) | (0.76, 0.96) | (0.85, 1.03) | |
| HR+C | 0.48 *,†,‡ | 0.20 | 0.41 | 0.29 |
| (0.09, 0.73) | (0.09, 0.60) | (0.12, 0.76) | (0.09, 0.67) |
| LDH Release 5 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 1.00 | 0.99 | 1.02 | 1.01 |
| (0.97, 1.03) | (0.97, 1.03) | (0.97, 1.05) | (0.97, 1.07) | |
| CN+C | 1.08 | 1.06 | 1.05 | 1.12 |
| (1.01, 1.14) | (1.01, 1.12) | (1.00, 1.09) | (1.04, 1.21) | |
| HR-C | 1.58 *,† | 1.58 | 1.54 | 1.53 |
| (1.41, 1.82) | (1.44, 1.78) | (1.36, 1.70) | (1.40, 1.70) | |
| HR+C | 1.58 4,*,† | 1.55 4 | 1.52 | 1.44 1,2 |
| (1.50, 1.80) | (1.47, 1.74) | (1.40, 1.76) | (1.33, 1.72) |
| Mitochondrial Activity 5 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 1.00 2,3,4 | 1.12 1 | 1.15 1 | 1.13 1 |
| (0.93, 1.07) | (0.97, 1.28) | (1.04, 1.32) | (1.01, 1.30) | |
| CN+C | 0.92 2,3,4 | 1.09 1 | 1.08 1 | 1.12 1 |
| (0.83, 1.08) | (1.00, 1.25) | (0.93, 1.36) | (0.95, 1.21) | |
| HR-C | 0.87 2,3,4,* | 0.96 1 | 1.02 1 | 1.04 1 |
| (0.71, 1.05) | (0.88, 1.15) | (0.87, 1.17) | (0.85, 1.23) | |
| HR+C | 0.70 2,3,4, *,†,‡ | 0.88 1 | 0.98 1 | 0.88 1 |
| (0.55, 0.86) | (0.73, 1.07) | (0.72, 1.15) | (0.68, 1.08) |
| Total NO Production 5 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 1.00 3 | 1.02 3 | 1.17 4 | 0.99 3 |
| (0.98, 1.02) | (0.94, 1.10) | (1.04, 1.30) | (0.94, 1.25) | |
| CN+C | 0.97 2,3,4 | 1.92 1 | 1.67 1,4 | 2.27 1,3 |
| (0.89, 1.29) | (1.64, 2.17) | (1.45, 1.83) | (2.10, 2.71) | |
| HR-C | 1.15 *,† | 1.19 | 1.15 | 1.20 |
| (1.07, 1.25) | (1.08, 1.28) | (1.08, 1.30) | (1.05, 1.28) | |
| HR+C | 2.10 *,†,‡ | 1.88 | 1.80 | 5.43 |
| (1.33, 3.95) | (1.45, 5.69) | (1.45, 6.17) | (3.99, 7.90) |
| Cell Number/Viability 15 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 0.98 2,4 | 0.93 1,3,4 | 0.98 2,4 | 1.04 1,2,3 |
| (0.94, 1.04) | (0.88, 0.96) | (0.91, 1.02) | (0.98, 1.10) | |
| CN+C | 0.74 | 0.35 | 0.45 | 0.46 |
| (0.52, 0.88) | (0.23, 0.66) | (0.11, 0.72) | (0.27, 0.76) | |
| HR-C | 0.04 4,*,† | 0.03 4 | 0.04 4 | 0.05 1,2,3 |
| (0.03, 0.05) | (0.03, 0.04) | (0.03, 0.05) | (0.04, 0.06) | |
| HR+C | 0.02 *,†,‡ | 0.02 | 0.02 | 0.03 |
| (0.01, 0.03) | (0.01, 0.04) | (0.02, 0.03) | (0.02, 0.04) |
| LDH Release 15 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 1.00 3 | 1.003 | 1.03 1,2 | 1.03 |
| (0.98, 1.04) | (0.97, 1.04) | (1.02, 1.07) | (1.01, 1.04) | |
| CN+C | 1.10 | 1.10 | 1.10 | 1.17 |
| (1.05, 1.13) | (1.05, 1.21) | (1.08, 1.19) | (1.06, 1.20) | |
| HR-C | 6.51 *,† | 6.30 | 6.32 | 6.03 |
| (5.41, 6.76) | (5.38, 6.48) | (5.09, 6.40) | (5.16, 6.32) | |
| HR+C | 5.41 *,† | 5.63 | 5.91 | 5.13 |
| (3.64, 7.05) | (4.14, 6.68) | (4.55, 6.65) | (3.59, 6.07) |
| Metabolic Activity 15 h P188 | 0 µM P188 1 | 10 µM P188 2 | 100 µM P188 3 | 1 mM P188 4 |
|---|---|---|---|---|
| CN-C | 1.00 2,3,4 | 1.11 1 | 1.24 1 | 1.21 1 |
| (0.92, 1.04) | (1.05, 1.25) | (1.14, 1.35) | (0.99, 1.46) | |
| CN+C | 0.85 2,3,* | 1.17 1 | 1.16 1 | 1.07 |
| (0.80, 0.91) | (1.03, 1.34) | (0.96, 1.29) | (0.88, 1.15) | |
| HR-C | 0.27 3,4,*,† | 0.26 3,4 | 0.45 1,2 | 0.46 1,2 |
| (0.21, 0.33) | (0.22, 0.42) | (0.33, 0.69) | (0.40, 0.57) | |
| HR+C | 0.33 3,4,*,† | 0.35 3 | 0.60 1,2 | 0.57 1 |
| (0.27, 0.39) | (0.26, 0.53) | (0.50, 0.68) | (0.48, 0.61) |
| Cell Number/Viability 5 h PEG | 0 µM PEG 1 | 10 µM PEG 2 | 100 µM PEG 3 | 1 mM PEG 4 |
|---|---|---|---|---|
| CN-C | 1.00 | 0.99 | 0.96 | 0.97 |
| (0.93, 1.07) | (0.94, 1.04) | (0.88, 1.04) | (0.87, 1.06) | |
| CN+C | 1.02 2,4 | 0.56 1 | 0.93 | 0.431 |
| (0.96, 1.14) | (0.46, 0.86) | (0.52, 1.12) | (0.24, 0.85) | |
| HR-C | 0.90 *,† | 0.90 | 0.93 | 0.94 |
| (0.81, 1.03) | (0.80, 1.00) | (0.84, 1.09) | (0.80, 1.05) | |
| HR+C | 0.55 *,†,‡ | 0.40 | 0.58 | 0.32 |
| (0.33, 0.69) | (0.15, 0.67) | (1.16, 0.73) | (0.06, 0.62) |
| LDH 5 h PEG | 0 µM PEG 1 | 10 µM PEG 2 | 100 µM PEG 3 | 1 mM PEG 4 |
|---|---|---|---|---|
| CN-C | 0.99 | 0.99 | 0.99 | 0.97 |
| (0.97, 1.06) | (0.94, 1.04) | (0.95, 1.02) | (0.93, 1.03) | |
| CN+C | 1.07 * | 1.08 | 1.04 | 1.05 |
| (1.03, 1.17) | (1.00, 1.13) | (0.96, 1.10) | (1.00, 1.12) | |
| HR-C | 1.32 *,† | 1.32 | 1.30 | 1.31 |
| (1.22, 1.42) | (1.25, 1.47) | (1.24, 1.41) | (1.24, 1.39) | |
| HR+C | 1.45 *,† | 1.35 | 1.35 | 1.32 |
| (1.32, 1.56) | (1.20, 1.45) | (1.27, 1.44) | (1.25, 1.43) |
| Metabolic Activity 5 h PEG | 0 µM PEG 1 | 10 µM PEG 2 | 100 µM PEG 3 | 1 mM PEG 4 |
|---|---|---|---|---|
| CN-C | 1.02 | 1.03 | 1.02 | 1.05 |
| (0.88, 1.09) | (0.96, 1.12) | (0.95, 1.15) | (0.90, 1.21) | |
| CN+C | 0.87 2,3,* | 1.05 1 | 1.12 1,4 | 0.94 3 |
| (0.77, 0.95) | (0.88, 1.20) | (0.87, 1.27) | (0.84, 1.02) | |
| HR-C | 0.75 2,3,4,* | 0.88 1 | 0.92 1 | 0.89 1 |
| (0.60, 0.93) | (0.66, 1.00) | (0.77, 1.02) | (0.74, 1.08) | |
| HR+C | 0.54 *,†,‡ | 0.72 | 0.73 | 0.73 |
| (0.48, 0.72) | (0.45, 0.85) | (0.53, 0.90) | (0.56, 0.88) |
References
- Mayer, A.R.; Quinn, D.K.; Master, C.L. The spectrum of mild traumatic brain injury: A review. Neurology 2017, 89, 623–632. [Google Scholar] [CrossRef]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef]
- Salehi, A.; Zhang, J.H.; Obenaus, A. Response of the cerebral vasculature following traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, K.; Unterberg, A. Schädel-Hirn-Trauma. Nervenarzt 2016, 87, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, P.C.; Welbourne, J.; Thomas, E.; Summers, F.; Whyte, M.; Hutchinson, P.J. Traumatic Brain Injury: A Multidisciplinary Approach, 2nd ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2020; ISBN 978-1-108-43086-9. [Google Scholar]
- Mazzeo, A.T.; Beat, A.; Singh, A.; Bullock, M.R. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp. Neurol. 2009, 218, 363–370. [Google Scholar] [CrossRef]
- Launey, Y.; Fryer, T.D.; Hong, Y.T.; Steiner, L.A.; Nortje, J.; Veenith, T.V.; Hutchinson, P.J.; Ercole, A.; Gupta, A.K.; Aigbirhio, F.I.; et al. Spatial and Temporal Pattern of Ischemia and Abnormal Vascular Function Following Traumatic Brain Injury. JAMA Neurol. 2020, 77, 339–349. [Google Scholar] [CrossRef]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. 2016, 5. [Google Scholar] [CrossRef]
- Kwiatkowski, T.A.; Rose, A.L.; Jung, R.; Capati, A.; Hallak, D.; Yan, R.; Weisleder, N. Multiple poloxamers increase plasma membrane repair capacity in muscle and nonmuscle cells. Am. J. Physiol. Cell Physiol. 2020, 318, C253–C262. [Google Scholar] [CrossRef]
- Wang, J.C.; Bindokas, V.P.; Skinner, M.; Emrick, T.; Marks, J.D. Mitochondrial mechanisms of neuronal rescue by F-68, a hydrophilic Pluronic block co-polymer, following acute substrate deprivation. Neurochem. Int. 2017, 109, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Goliaei, A.; Tsereteli, L.; Berkowitz, M.L. Properties of Poloxamer Molecules and Poloxamer Micelles Dissolved in Water and Next to Lipid Bilayers: Results from Computer Simulations. J. Phys. Chem. B 2016, 120, 5823–5830. [Google Scholar] [CrossRef] [PubMed]
- Salzman, M.M.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Protects Isolated Adult Mouse Cardiomyocytes from Reoxygenation Injury. Pharmacol. Res. Perspect. 2020, 8, e00639. [Google Scholar] [CrossRef] [PubMed]
- Salzman, M.M.; Bates, F.S.; Hackel, B.J.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Membrane Repair by Poloxamer 188 Protects Isolated Cardiac Cells From Hypoxia/Reoxygenation Injury. Circulation 2017, 136, A18264. [Google Scholar]
- Serbest, G.; Horwitz, J.; Barbee, K. The effect of poloxamer-188 on neuronal cell recovery from mechanical injury. J. Neurotrauma 2005, 22, 119–132. [Google Scholar] [CrossRef]
- Serbest, G.; Horwitz, J.; Jost, M.; Barbee, K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006, 20, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D.; Gallo, G.; Barbee, K. Poloxamer 188 reduces axonal beading following mechanical trauma to cultured neurons. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2007, 2007, 5395–5398. [Google Scholar] [CrossRef]
- Luo, C.-L.; Chen, X.-P.; Li, L.-L.; Li, Q.-Q.; Li, B.-X.; Xue, A.-M.; Xu, H.-F.; Dai, D.-K.; Shen, Y.-W.; Tao, L.-Y.; et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J. Neurotrauma 2013, 30, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Inyang, E.; Abhyankar, V.; Chen, B.; Cho, M. Modulation of in vitro Brain Endothelium by Mechanical Trauma: Structural and Functional Restoration by Poloxamer 188. Sci. Rep. 2020, 10, 3054. [Google Scholar] [CrossRef]
- Kanagaraj, J.; Chen, B.; Xiao, S.; Cho, M. Reparative Effects of Poloxamer P188 in Astrocytes Exposed to Controlled Microcavitation. Ann. Biomed. Eng. 2018, 46, 354–364. [Google Scholar] [CrossRef]
- Gu, J.-H.; Ge, J.-B.; Li, M.; Xu, H.-D.; Wu, F.; Qin, Z.-H. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE 2013, 8, e61641. [Google Scholar] [CrossRef]
- Goliaei, A.; Lau, E.Y.; Adhikari, U.; Schwegler, E.; Berkowitz, M.L. Behavior of P85 and P188 Poloxamer Molecules: Computer Simulations Using United-Atom Force-Field. J. Phys. Chem. B 2016, 120, 8631–8641. [Google Scholar] [CrossRef]
- Bao, H.-J.; Wang, T.; Zhang, M.-Y.; Liu, R.; Dai, D.-K.; Wang, Y.-Q.; Wang, L.; Zhang, L.; Gao, Y.-Z.; Qin, Z.-H.; et al. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem. Res. 2012, 37, 2856–2867. [Google Scholar] [CrossRef]
- Wang, T.; Chen, X.; Wang, Z.; Zhang, M.; Meng, H.; Gao, Y.; Luo, B.; Tao, L.; Chen, Y. Poloxamer-188 can attenuate blood-brain barrier damage to exert neuroprotective effect in mice intracerebral hemorrhage model. J. Mol. Neurosci. 2015, 55, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Lotze, F.P.; Salzman, M.M.; Pille, J.A.; Balzer, C.; Hees, J.E.; Cleveland, W.J.; Riess, M.L. Poloxamer 188 Protects Mouse Brain Microvascular Endothelial Cells in an in-vitro Traumatic Brain Injury Model. Circulation 2019, 140, A22. [Google Scholar] [CrossRef]
- Lotze, F.P.; Salzman, M.M.; Pille, J.A.; Balzer, C.; Hees, J.E.; Cleveland, W.; Riess, M.L. In Vitro Traumatic Brain Injury Model of Mouse Brain Microvascular Endothelial Cells. FASEB J. 2019, 33, 527.4. [Google Scholar] [CrossRef]
- Meyer, L.J.; Riess, M.L. Evaluation of In Vitro Neuronal Protection by Postconditioning with Poloxamer 188 Following Simulated Traumatic Brain Injury. Life 2021, 11, 316. [Google Scholar] [CrossRef] [PubMed]
- Barltrop, J.A.; Owen, T.C.; Cory, A.H.; Cory, J.G. 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans As cell-viability indicators. Bioorganic Med. Chem. Lett. 1991, 1, 611–614. [Google Scholar] [CrossRef]
- Boron, W.F.; Boulpaep, E.L. Medical Physiology, 3rd ed.; Elsevier Health Sciences: London, UK, 2017; ISBN 978-1-4557-4377-3. [Google Scholar]
- Centers for Disease Control and Prevention. Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths-United States, 2014; Centers for Disease Control and Prevention, U.S. Department of Health and Human Services: Washington, DC, USA, 2019. [Google Scholar]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Murphy, A.D.; McCormack, M.C.; Bichara, D.A.; Nguyen, J.T.; Randolph, M.A.; Watkins, M.T.; Lee, R.C.; Austen, W.G. Poloxamer 188 protects against ischemia-reperfusion injury in a murine hind-limb model. Plast. Reconstr. Surg. 2010, 125, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D.; Gallo, G.; Barbee, K.A. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp. Neurol. 2008, 212, 422–430. [Google Scholar] [CrossRef]
- Luo, C.; Li, Q.; Gao, Y.; Shen, X.; Ma, L.; Wu, Q.; Wang, Z.; Zhang, M.; Zhao, Z.; Chen, X.; et al. Poloxamer 188 Attenuates Cerebral Hypoxia/Ischemia Injury in Parallel with Preventing Mitochondrial Membrane Permeabilization and Autophagic Activation. J. Mol. Neurosci. 2015, 56, 988–998. [Google Scholar] [CrossRef]
- Shelat, P.B.; Plant, L.D.; Wang, J.C.; Lee, E.; Marks, J.D. The membrane-active tri-block copolymer pluronic F-68 profoundly rescues rat hippocampal neurons from oxygen-glucose deprivation-induced death through early inhibition of apoptosis. J. Neurosci. 2013, 33, 12287–12299. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Emanuele, M.; Zhang, L.; Zhang, Z.G.; Lu, M.; Zhang, T.; Mahmood, A.; Xiong, Y. Treatment of Traumatic Brain Injury with Vepoloxamer (Purified Poloxamer 188). J. Neurotrauma 2018, 35, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Cadichon, S.B.; Le Hoang, M.; Wright, D.A.; Curry, D.J.; Kang, U.; Frim, D.M. Neuroprotective effect of the surfactant poloxamer 188 in a model of intracranial hemorrhage in rats. J. Neurosurg. 2007, 106, 36–40. [Google Scholar] [CrossRef]
- Marks, J.D.; Pan, C.Y.; Bushell, T.; Cromie, W.; Lee, R.C. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001, 15, 1107–1109. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Poloxamer 188 volumetrically decreases neuronal loss in the rat in a time-dependent manner. Neurosurgery 2004, 55, 943–948. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Surfactant poloxamer 188-related decreases in inflammation and tissue damage after experimental brain injury in rats. J. Neurosurg. 2004, 101, 91–96. [Google Scholar] [CrossRef]
- Frim, D.M.; Wright, D.A.; Curry, D.J.; Cromie, W.; Lee, R.; Kang, U.J. The surfactant poloxamer-188 protects against glutamate toxicity in the rat brain. Neuroreport 2004, 15, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Kondaveeti, P.; Nissanov, J.; Barbee, K.; Shewokis, P.; Rioux, L.; Moxon, K.A. Preventing neuronal damage and inflammation in vivo during cortical microelectrode implantation through the use of poloxamer P-188. J. Neural Eng. 2013, 10, 16011. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Lin, H.; Hong, X.; Ji, D.; Wu, F. Poloxamer 188-mediated anti-inflammatory effect rescues cognitive deficits in paraquat and maneb-induced mouse model of Parkinson’s disease. Toxicology 2020, 436, 152437. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef]
- Colbassani, H.J.; Barrow, D.L.; Sweeney, K.M.; Bakay, R.A.E.; Check, I.J.; Hunter, R.L. Modification of acute focal ischemia in rabbits by poloxamer 188. Stroke 1989, 20, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Sigma-Aldrich. Poly(Ethylene Glycol) Average Mol Wt 8000. Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/p2139?lang=de®ion=DE (accessed on 2 June 2021).
- Sigma-Aldrich. Kolliphor®P 188. Available online: https://www.sigmaaldrich.com/catalog/substance/kolliphorp18812345900311611?lang=de®ion=DE (accessed on 2 June 2021).
- Maskarinec, S.A.; Hannig, J.; Lee, R.C.; Lee, K.Y.C. Direct Observation of Poloxamer 188 Insertion into Lipid Monolayers. Biophys. J. 2002, 82, 1453–1459. [Google Scholar] [CrossRef]
- Maskarinec, S.A.; Wu, G.; Lee, K.Y.C. Membrane sealing by polymers. Ann. N. Y. Acad. Sci. 2005, 1066, 310–320. [Google Scholar] [CrossRef]
- Zhao, M.; Thuret, G.; Piselli, S.; Pipparelli, A.; Acquart, S.; Peoc’h, M.; Dumollard, J.-M.; Gain, P. Use of poloxamers for deswelling of organ-cultured corneas. Investig. Ophthalmol. Vis. Sci. 2008, 49, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qin, Y.; Huang, Y.; Ji, D.; Wu, F. Poloxamer 188 rescues MPTP-induced lysosomal membrane integrity impairment in cellular and mouse models of Parkinson’s disease. Neurochem. Int. 2019, 126, 178–186. [Google Scholar] [CrossRef]
- Follis, F.; Jenson, B.; Blisard, K.; Hall, E.; Wong, R.; Kessler, R.; Temes, T.; Wernly, J. Role of poloxamer 188 during recovery from ischemic spinal cord injury: A preliminary study. J. Investig. Surg. 1996, 9, 149–156. [Google Scholar] [CrossRef]
- Martindale, J.J.; Metzger, J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J. Mol. Cell. Cardiol. 2014, 67, 26–37. [Google Scholar] [CrossRef]
- Huang, L.; Perrault, C.; Coelho-Martins, J.; Hu, C.; Dulong, C.; Varna, M.; Liu, J.; Jin, J.; Soria, C.; Cazin, L.; et al. Induction of acquired drug resistance in endothelial cells and its involvement in anticancer therapy. J. Hematol. Oncol. 2013, 6, 49. [Google Scholar] [CrossRef]
- Tskitishvili, E.; Sharentuya, N.; Temma-Asano, K.; Mimura, K.; Kinugasa-Taniguchi, Y.; Kanagawa, T.; Fukuda, H.; Kimura, T.; Tomimatsu, T.; Shimoya, K. Oxidative stress-induced S100B protein from placenta and amnion affects soluble Endoglin release from endothelial cells. Mol. Hum. Reprod. 2010, 16, 188–199. [Google Scholar] [CrossRef]
- CyQUANT Direct Cell Proliferation Assay Kit. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2Fmp35011.pdf&title=Q3lRVUFOVCBEaXJlY3QgQ2VsbCBQcm9saWZlcmF0aW9uIEFzc2F5IEtpdA== (accessed on 23 May 2021).
- CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS). Available online: https://www.promega.de/products/cell-health-assays/cell-viability-and-cytotoxicity-assays/celltiter-96-aqueous-one-solution-cell-proliferation-assay-_mts_/?catNum=G3582 (accessed on 23 May 2021).
- Bartos, J.A.; Matsuura, T.R.; Tsangaris, A.; Olson, M.; McKnite, S.H.; Rees, J.N.; Haman, K.; Shekar, K.C.; Riess, M.L.; Bates, F.S.; et al. Intracoronary Poloxamer 188 Prevents Reperfusion Injury in a Porcine Model of ST-Segment Elevation Myocardial Infarction. JACC Basic Transl. Sci. 2016, 1, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Cherian, L.; Hlatky, R.; Robertson, C.S. Nitric Oxide in Traumatic Brain Injury. Brain Pathol. 2004, 14, 195–201. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.B.; Pan, Z.H.; Lei, S.Z.; Chen, H.S.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Zweckberger, K.; Trabold, R.; Schilling, L.; Schinzel, R.; Tegtmeier, F.; Plesnila, N. The novel nitric oxide synthase inhibitor 4-amino-tetrahydro-L-biopterine prevents brain edema formation and intracranial hypertension following traumatic brain injury in mice. J. Neurotrauma 2009, 26, 1963–1975. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The role of the nitric oxide pathway in brain injury and its treatment--from bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Douglas, H.F.; Salzman, M.M.; Hackel, B.J.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Dose Dependent Nitric Oxide Production by Poloxamer 188 in Rat Isolated Hearts. FASEB J. 2018, 31, 1006-3. [Google Scholar] [CrossRef]
- Salzman, M.M.; Cheng, Q.; Matsuura, T.R.; Yannopoulos, D.; Riess, M.L. Cardioprotection by Poloxamer 188 is Mediated by Nitric Oxide Synthase. FASEB J. 2015, 29, 1026.5. [Google Scholar] [CrossRef]
- Villalba, N.; Sackheim, A.M.; Nunez, I.A.; Hill-Eubanks, D.C.; Nelson, M.T.; Wellman, G.C.; Freeman, K. Traumatic Brain Injury Causes Endothelial Dysfunction in the Systemic Microcirculation through Arginase-1-Dependent Uncoupling of Endothelial Nitric Oxide Synthase. J. Neurotrauma 2017, 34, 192–203. [Google Scholar] [CrossRef]
- Lucas, R.; Fulton, D.; Caldwell, R.W.; Romero, M.J. Arginase in the vascular endothelium: Friend or foe? Front. Immunol. 2014, 5, 589. [Google Scholar] [CrossRef][Green Version]
- Igarashi, J.; Michel, T. S1P and eNOS regulation. Biochim. Biophys. Acta 2008, 1781, 489–495. [Google Scholar] [CrossRef]
- Morrison, B.; Elkin, B.S.; Dollé, J.-P.; Yarmush, M.L. In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 2011, 13, 91–126. [Google Scholar] [CrossRef]
- Song, H.W.; Foreman, K.L.; Gastfriend, B.D.; Kuo, J.S.; Palecek, S.P.; Shusta, E.V. Transcriptomic comparison of human and mouse brain microvessels. Sci. Rep. 2020, 10, 12358. [Google Scholar] [CrossRef]
- Tsukada, Y.; Muramatsu, F.; Hayashi, Y.; Inagaki, C.; Su, H.; Iba, T.; Kidoya, H.; Takakura, N. An in vivo model allowing continuous observation of human vascular formation in the same animal over time. Sci. Rep. 2021, 11, 745. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hampton, M.J.W.; Barajas, M.B.; Riess, M.L. Spatial Distance Affects Hypoxia/Reoxygenation Injury in a Co-Culture of Separate Endothelial Cell and Cardiomyocyte Layers. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 523–534. [Google Scholar] [CrossRef]
- Datta, D.; Vasudevan, A. Migration, Chemo-Attraction, and Co-Culture Assays for Human Stem Cell-Derived Endothelial Cells and GABAergic Neurons. J. Vis. Exp. 2020, 155. [Google Scholar] [CrossRef] [PubMed]
- Moysidou, C.-M.; Barberio, C.; Owens, R.M. Advances in Engineering Human Tissue Models. Front. Bioeng. Biotechnol. 2020, 8, 620962. [Google Scholar] [CrossRef]
- Faley, S.L.; Neal, E.H.; Wang, J.X.; Bosworth, A.M.; Weber, C.M.; Balotin, K.M.; Lippmann, E.S.; Bellan, L.M. iPSC-Derived Brain Endothelium Exhibits Stable, Long-Term Barrier Function in Perfused Hydrogel Scaffolds. Stem Cell Rep. 2019, 12, 474–487. [Google Scholar] [CrossRef]
- Houang, E.M.; Haman, K.J.; Kim, M.; Zhang, W.; Lowe, D.A.; Sham, Y.Y.; Lodge, T.P.; Hackel, B.J.; Bates, F.S.; Metzger, J.M. Chemical End Group Modified Diblock Copolymers Elucidate Anchor and Chain Mechanism of Membrane Stabilization. Mol. Pharm. 2017, 14, 2333–2339. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lotze, F.P.; Riess, M.L. Poloxamer 188 Exerts Direct Protective Effects on Mouse Brain Microvascular Endothelial Cells in an In Vitro Traumatic Brain Injury Model. Biomedicines 2021, 9, 1043. https://doi.org/10.3390/biomedicines9081043
Lotze FP, Riess ML. Poloxamer 188 Exerts Direct Protective Effects on Mouse Brain Microvascular Endothelial Cells in an In Vitro Traumatic Brain Injury Model. Biomedicines. 2021; 9(8):1043. https://doi.org/10.3390/biomedicines9081043
Chicago/Turabian StyleLotze, Felicia P., and Matthias L. Riess. 2021. "Poloxamer 188 Exerts Direct Protective Effects on Mouse Brain Microvascular Endothelial Cells in an In Vitro Traumatic Brain Injury Model" Biomedicines 9, no. 8: 1043. https://doi.org/10.3390/biomedicines9081043
APA StyleLotze, F. P., & Riess, M. L. (2021). Poloxamer 188 Exerts Direct Protective Effects on Mouse Brain Microvascular Endothelial Cells in an In Vitro Traumatic Brain Injury Model. Biomedicines, 9(8), 1043. https://doi.org/10.3390/biomedicines9081043