1. Introduction
Fibroblast growth factor receptors (FGFRs) are important members of the tyrosine kinase receptor family [
1]. The FGFR family contains four members: FGFR1, FGFR2, FGFR3 and FGFR4 [
2]. Most of them are single-chain glycoprotein molecules, which are divided into the extracellular region, transmembrane region and intracellular region [
3]. Under normal physiological conditions, an FGFR binds to its ligand fibroblast growth factor (FGF). The FGFR dimer and self-phosphorylation activate downstream signaling pathways such as the JAK/STAT pathway and phospholipase C pathway, which play important roles in tumor growth and angiogenesis [
4]. Abnormalities in FGFR are closely related to the occurrence of many types of tumors, such as lung cancer, breast cancer, gastric cancer, etc. Therefore, FGFR-targeting drugs can have direct or indirect antitumor effects [
5,
6].
Erdafitinib is the first FDA-approved FGFR tyrosine kinase inhibitor for treating urothelial carcinoma [
7]. There are currently 87 FGFR-targeting drugs at the clinical stage. These drugs are mainly used in the field of tumor treatment, such as infratinib, pemigatinib, rogaratinib, AZD-4547, PRN-1371 and mastinib mesylate. Among them, seven drugs are in clinical phase III [
8]. It is expected that more FGFR drugs will enter the market in the next few years. Therefore, the competition for FGFR-target inhibitors is high.
An antibody–drug conjugate (ADC) is a monoclonal antibody coupled with a cytotoxin through a specific linker, which combines the high selectivity of antibodies with an antitumor toxin. Its overall structure can be divided into three different structural modules: an antibody, cytotoxic compound and linker [
9,
10]. Compared with traditional chemotherapy drugs, an ADC has stronger killing effects on cancer cells. It exerts activity at pmol and nmol concentrations and should not exhibit immunogenicity after entering the human body [
11,
12]. Ten ADC drugs have been approved by the FDA to date. Among them, Polivy, Padcev, Enthertu and Trodelvy had just been approved in the previous year at the time of writing. This indicates a breakthrough in this development field.
Compared with ADCs, peptide–drug conjugates (PDCs) are in the development stage. Similarly, a PDC is a target peptide ligand conjugated to a cytotoxin via a cleavable linker. PDCs have many advantages, such as small size, excellent cell permeability and high drug-loading capability [
13]. However, how to obtain an efficient target peptide to specifically recognize the tumor is very important for PDC development.
The OBOC combination peptide library was developed more than two decades ago, which can generate thousands to millions of resin beads containing compounds for rapidly screening a peptide, and each bead contains only one compound [
14]. Compared to phage-display methods, we can obtain peptides containing unnatural amino acid residues by using the OBOC library. The obtained peptides have high stability because they are not easily recognized and degraded by proteases [
15]. Moreover, the OBOC technology offers many more structural possibilities, e.g., linear, cyclic, branched and macrocyclic peptide libraries, which are typically synthesized on solid-phase resin beads using fmoc chemistry and split-mix synthesis [
16].
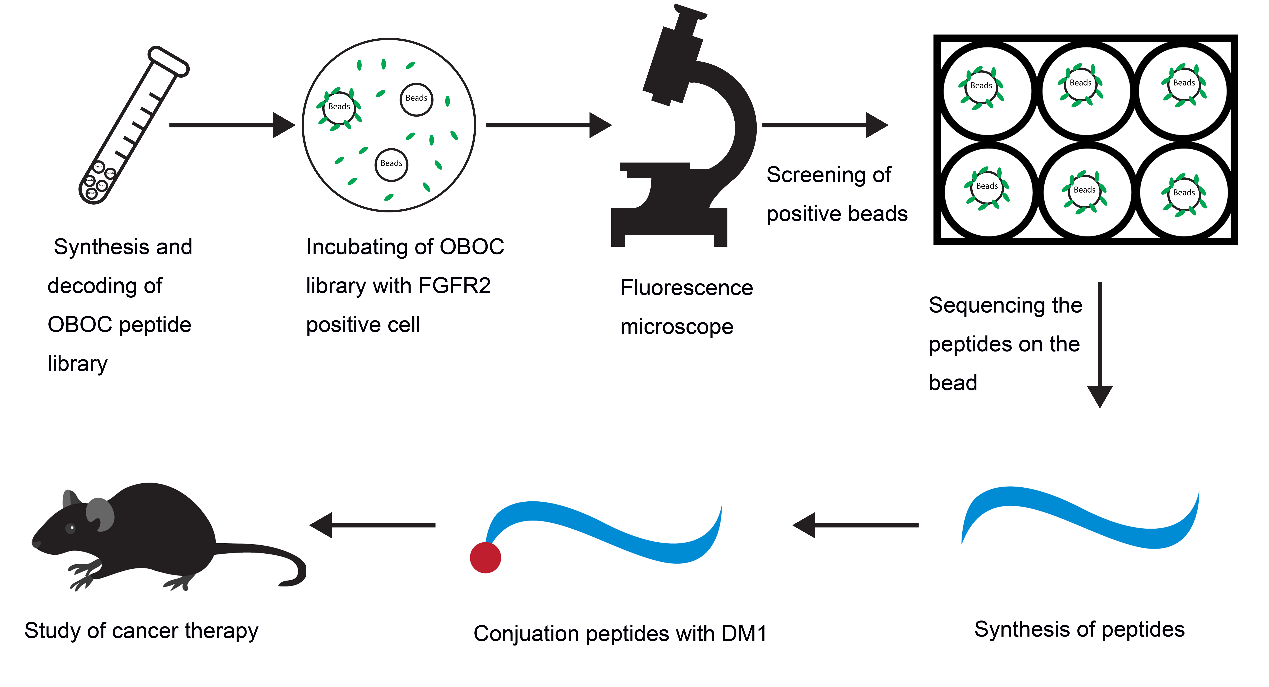
In this study, we tried to develop novel PDCs with high affinity for FGFR2 for tumor treatment. Using OBOC technology and FGFR2-positive cells, we screened and obtained a specific peptide named LLC2B, with 8 unnatural amino acid residues. Then, we coupled the LLC2B peptide with the cytotoxin DM1 to form a peptide–drug conjugate, which has an obvious inhibitory effect on tumor proliferation in vitro and in vivo.
2. Materials and Methods
2.1. Materials
The peptides named LLC2A, LLC2B, LLC2C, LLC2D, LLC2E and LLC2F were all synthesized by Shanghai Qiangyao Biological Technology Co., Ltd., Shanghai, China. The OBOC peptide library and PDC were synthesized by the Kit Lam laboratory, UC Davis, United States. The linking molecules Fmoc-Lys (Dde)-OH and Fmoc-AEEA, which were for drug synthesis, were purchased from ChemPep Company, Palm Beach, FL, USA; amide resin was purchased from P3BioSystems Co., Ltd., Louisville, KY, USA; HCTU and 3-(2-pyridyldithio)propionic acid were purchased from Matrix Scientific Co., Ltd., Elgin, SC, USA; 6-Cl HOBt and natural and unnatural amino acids were all purchased from Chem-Impex Co., Ltd., Bensenville, IL, USA; fluorescein-labeled Cyanine5.5-NHS was purchased from LuMiprobe Co., Ltd.; 4-methylpiperidine was purchased from Alfa Aesar Co., Ltd., Ward Hill, MA, USA; DIEA, trifluoroacetic acid, anisole, DIC and other organic solvents were all purchased from Sigma-Aldrich Co., Ltd., Saint Louis, MO, USA; DM1 was purchased from BoRui Co., Ltd., SuZhou, China. FGFR2 was purchased from Sino Biological Co., Ltd., Beijing, China; anti-FGFR2 (Bek(C-17):sc-122) was acquired from Santa Cruz Blotechnology, Santa Cruz, CA, USA.
2.2. Cells
The HEK293, HUVEC, MCF-7, EC109, KYSE180 and KYSE510 cell lines were purchased from the American Type Culture Collection (ATCC). All the cells were cultured in RPMI-1640 Medium (Gibco, USA), which was supplemented with 10% fetal bovine serum (Biological Industrie, Israel). The cell lines were incubated in a humidified atmosphere with 5% CO2 at 37 °C.
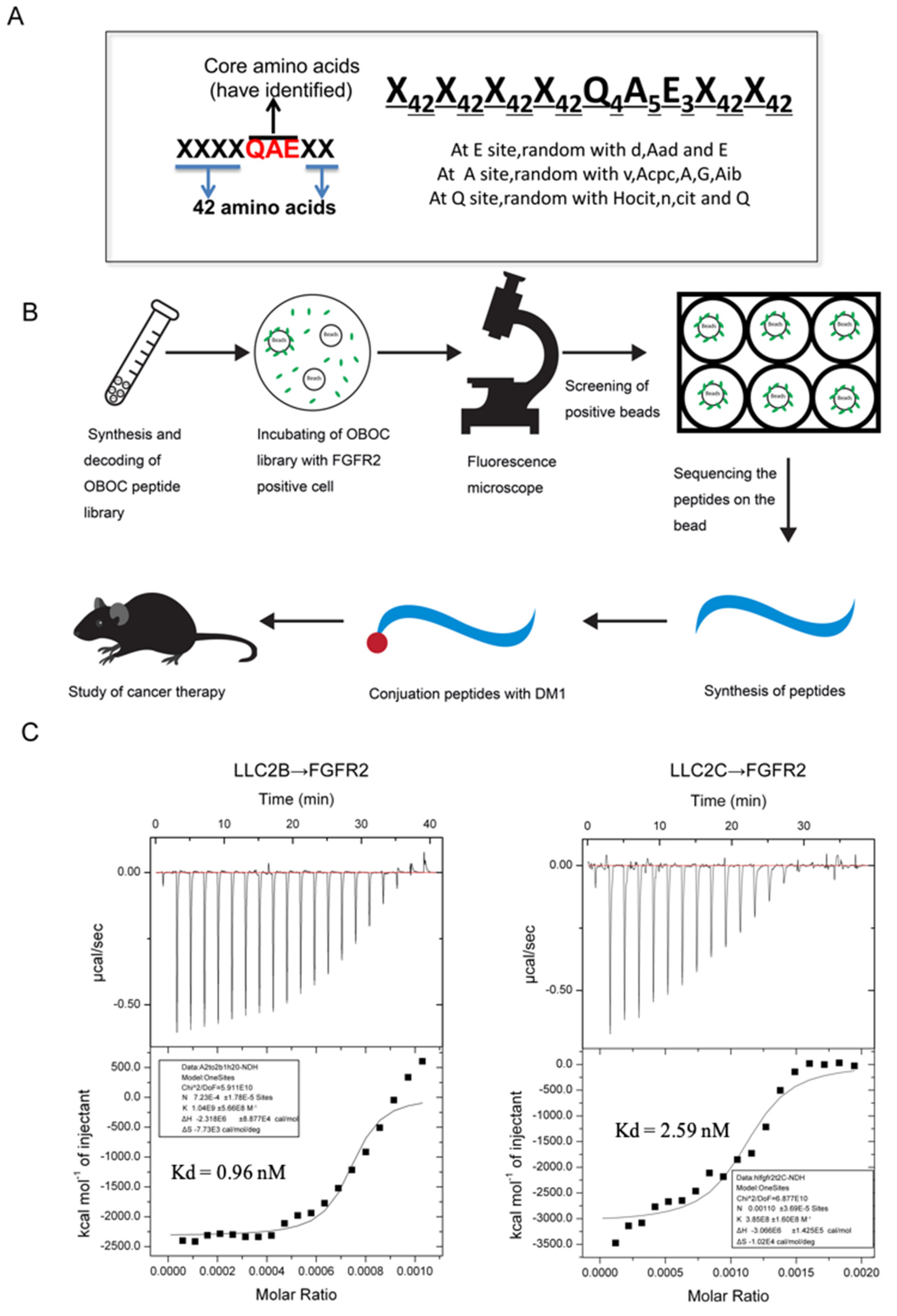
2.3. Procedure for the Identification of FGFR2-Binding Peptides
DMF (90%) was used to suspend the OBOC library beads and place them into 6-well plates with even spreading. The plate was not moved, and the beads were allowed to attach to the bottom of the plate for 1.5–2 h. Then, the 90% DMF was discarded, and the beads were washed once with 50% DMF. Then, the beads were washed with sterile water three times and PBS twice in a clean cell culture hood. Next, 293 and 293-FGFR2 cells were collected in sterile tubes with complete medium (CM). The cell density was adjusted to 1 × 10
6 per mL with CM. First, 1 mL of CM was added into each well with beads, and then, 500 µL of 293 cells and 500 µL of 293-FGFR2
+ cells were added. In total, there were 1 × 10
6 cells in 2 mL of CM per well. The plate was placed in a 37 °C, 5% CO
2 incubator with gentle shaking for 1 h. The CM was discarded, the cells were washed with PBS, and PBS was added to cover the cells. The wells were observed under a fluorescent microscope to pick out the beads binding in the 293-FGFR2 cells with GFP expression. The selected beads were put into PBS buffer, and then, 8 M guanidine hydrochloride was used to wash away attached cells. Consequently, the peptides on the isolated beads were sequenced using an amino acid sequencer. The peptides were then synthesized according to the results of the sequencing. The sequences of the peptides are shown in
Supplementary Table S2.
2.4. The Synthesis of LLC2B-Mal-DM1 and LLC2B-SS-DM1
First, the protected LLC2B-Lys (Dde) was assembled on rink amide resin using HCTU/DIEA activation for Fmoc/t-Bu chemistry. Then, half of the protected LLC2B-Lys (Dde) beads were transferred to a 10-mL polypropylene column with a frit. The Dde protecting group was removed with 2% NH2NH2 in DMF twice (5 min and 10 min). The beads were washed with DMF, MeOH and DMF, three times with each agent. 3-Maleimidopropionic acid (213 mg, 1.26 mmoL, 5 eq. to beads), 6-Cl HOBt (213 mg, 1.26 mmoL), and DIC (195 µL, 1.26 mmoL) were dissolved in DMF (6 mL) and mixed for 15 min before being added to the beads. The coupling reaction was conducted at room temperature for 2 h to yield protected LLC2B-K (Mal) beads. After the liquid was drained by vacuum, the beads were thoroughly washed with DMF, MeOH and dichloromethane (DCM), three times with each, and then dried under vacuum. The LLC2B-K (Mal) was cleaved off the beads with a TFA cocktail containing TFA (87.5%)/thioanisole (5%)/triisopropylsilane (2.5%)/water (5%) for 3 h. The liquid was collected and precipitated with cold ethyl ether. The precipitate was washed two more times with cold ethyl ether and dried over vacuum. LLC2B-K(Mal) (150 mg, 0.06 mmoL) powder was weighed out and dissolved in a mixture of acetonitrile (ACN) and water (1:1), and then added to PBS buffer (pH 7.2) containing 10 mM EDTA; then, through HCl and NaOH adjusted the pH to 7.0. A solution of DM1 (40 mg, 0.054 mmoL) in ACN was slowly added to the solution of LLC2B-K (Mal), which was stirred at room temperature for 1 h, until an Ellman test was negative. The resulting solution was directly used for reversed-phase HPLC purification on a preparative Vydac C18 column (22 × 250 mm) with a flow rate of 5 mL/min. The synthesis approach for LLC2B-SS-DM1 is similar to this method. We just replaced the 3-nmaleimidopropionic acid with 3-(2-pyridyldithio) propionic acid.
2.5. The Synthesis of LLC2B-Cy5.5 and S-LLC2B-Cy5.5
A portion of the protected-LLC2B-Lys (Dde) beads (~50 mg) was transferred to a 1.5 mL polypropylene column with a frit. After the Dde beads were removed and thoroughly wash as indicated above, Cyanine5.5-NHS (38 mg, 0.05 mmoL) and DIEA (17.5 µL, 0.1 mmoL) in anhydrous DMF (0.7 mL) were added to the beads, which were then incubated with rotation at room temperature in the dark (column wrapped with foil) for 2 h. The crude product was cleaved off the beads using a TFA cocktail as described above and precipitated with cold ethyl ether. HPLC purification yielded LLC2B-Cyanine5.5 as a dark blue powder (purity > 95%). The identity was confirmed by MALDI-TOF MS on a BRUKER microflex (ion positive reflector mode). MALDI-TOF MS Calculated: 3111.67. Found: 3112.21. The synthesis of S-LLC2B-Cyanine5.5 is similar to that of LLC2B-Cyanine5.5, except that the amino acid sequence is (hydroxyproline)-(D-Leu)-(citrulline)-(D-Asp)-(D-Asn)-(D-Lys)-(1-aminocyclopropanecarboxylic acid)-(norvaline)-(2-aminoadipic acid), the structure of amino acids is shown in
Table S1.
2.6. Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out with an ITC200 microcalorimeter (MicroCal, Northampton, MA, USA) as described previously [
17]. Both the LLC2B peptide and the recombinant proteins of the FGFR2 (Sino Biological, Beijing, China) were dissolved in sterile water. These samples were thoroughly degassed and then centrifuged to remove precipitates. The binding of both was measured by ITC at 25 °C using 0.1 mM peptide in the sample cell and 0.001 mM FGFR protein in the injecting syringe. Throughout the experiment, 19 drops were injected into the sample cell, and each drop had a volume of 2 μL. The interval between each drop was 5 min. Origin 7 software (Northampton, MA, USA) was applied for evaluating the experimental raw ITC data.
2.7. Immunofluorescence
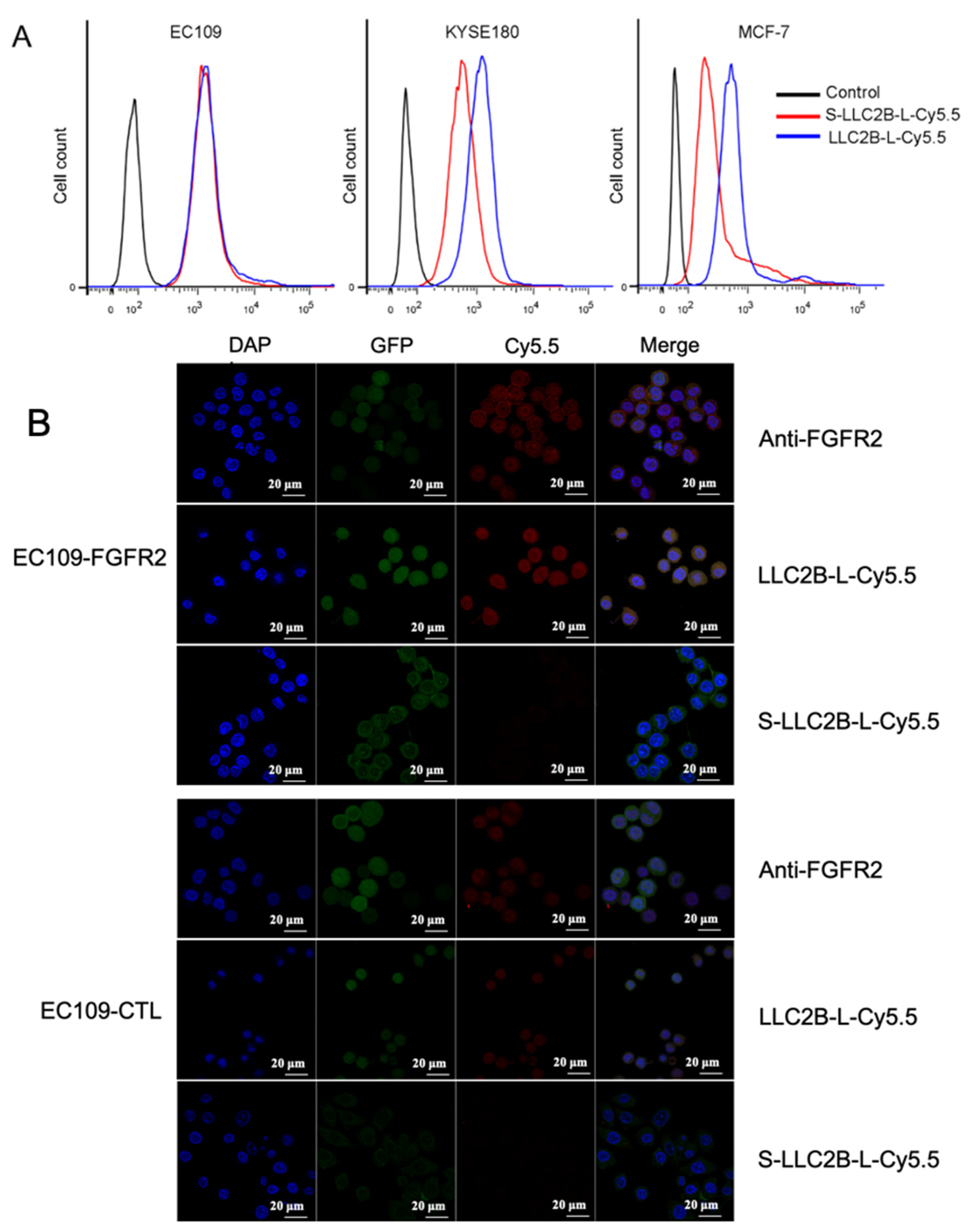
For in vitro immunofluorescence analysis, cells were fixed in 4% paraformaldehyde at room temperature for 15 min and then washed 3 times for 5 min each with PBS. Subsequently, the cells were incubated for 10 min in permeabilization solution (PBS; 0.25% Triton X-100) and then washed again with PBS 3 times for 5 min each. The cells were blocked in blocking solution (PBS; 1% BSA; 0.1% Tween 20) for 30 min, incubated overnight at 4 °C with primary antibodies, anti-FGFR2 (Bek(C-17):sc-122) in blocking solution, and washed intensively 5 times for 5 min each with PBST. Cy5.5-labeled secondary antibody was then applied for 1 h at room temperature, following which the cells were stained with DAPI (staining of nuclei) for 10 min. The images were acquired on a confocal microscope (Zeiss LSM 700, Germany).
2.8. In Vivo Targeting of LLC2B
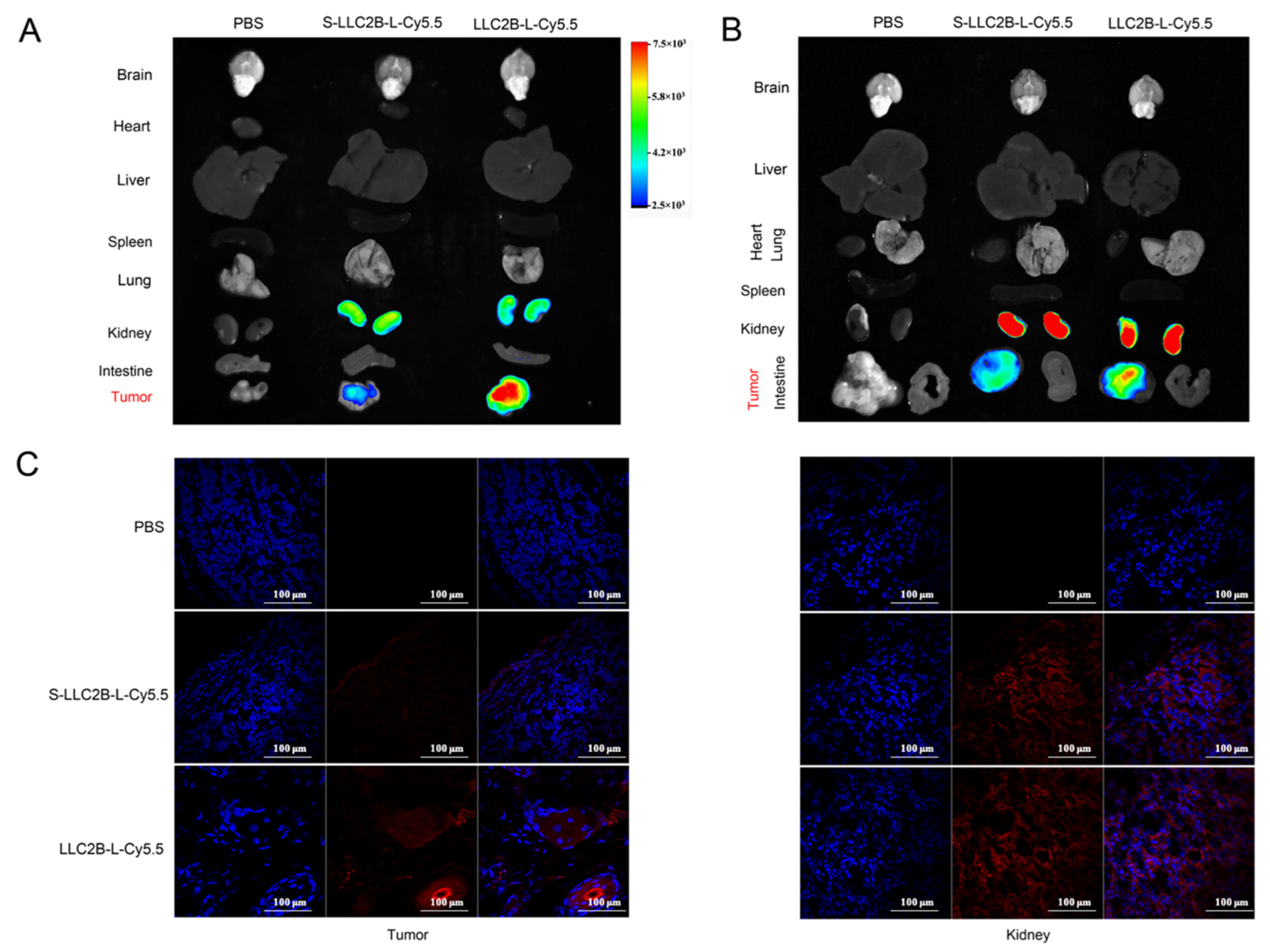
PBS, S-LLC2B-L-Cy5.5 (40 nmoL) and LLC2B-L-Cy5.5 (40 nmoL) were injected intravenously into MCF-7 or KYSE180 tumor-bearing nude mice. After 24 h, the Cy5.5 fluorescence in the entire body of the mouse was acquired by using an In-Vivo Xtreme (Bruker, Germany). After the mouse was sacrificed, the Cy5.5 fluorescence in the tumor, brain, liver, heart, spleen and lung tissues was quantified. The radiant efficiency was measured using the MI SE software (Bruker, Germany) and normalized by tissue volume.
2.9. Peptide Stability Testing In Vitro
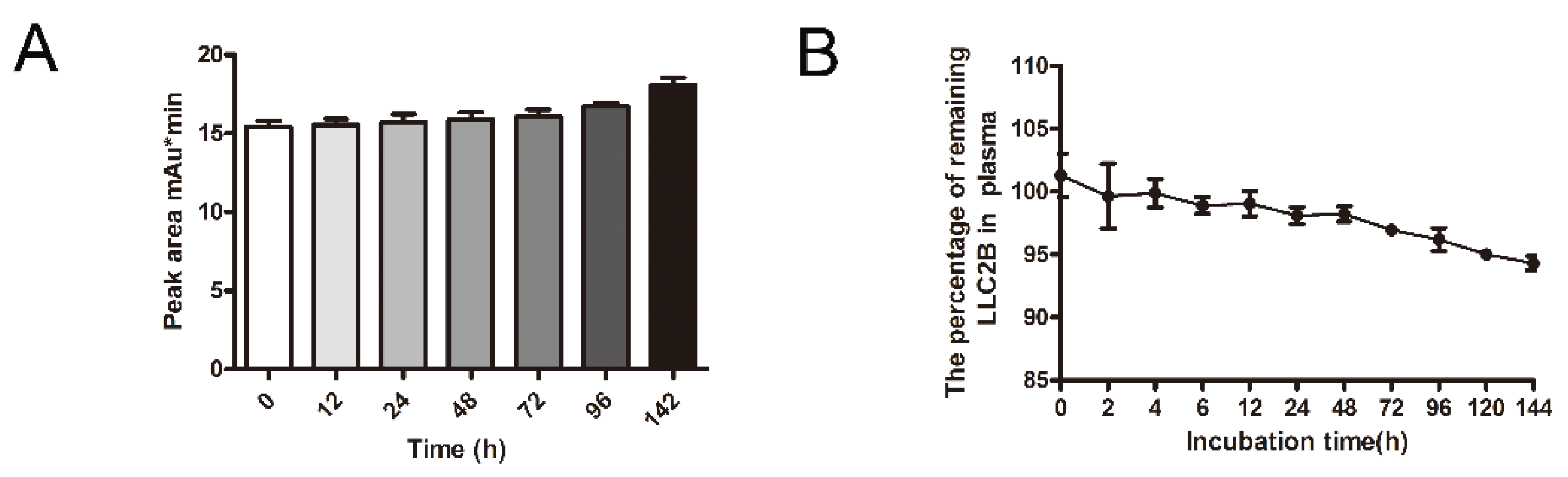
First, 9 mL of human plasma was mixed with 1 mL of LLC2B, with a final LLC2B concentration of 100 µM, and the mixture was incubated at 37 °C. Then, 250 μL of the mixture was removed after 0, 2, 4, 6, 12, 24, 48, 72, 96, 120 and 144 h of incubation, into 3 tubes each time. Next, 100 µL of TFA added, and the mixture was vortexed for 30 s, placed at room temperature for 10–30 min, and centrifuged at 13,000× g rpm/min for 15 min. The supernatant was removed and centrifuged for 5 min, and then, the new supernatant was taken for analysis. The peak area method was used for quantitative detection, and the peak area was used to generate a standard curve for obtaining the polypeptide concentrations at the time points. A similar method was used to measure the stability at room temperature. The peptide was placed at room temperature without plasma incubation, and samples were taken at 0, 12, 24, 48, 72, 96 and 142 h and used for high-performance liquid chromatography (HPLC) (ThermoFly, Ultimate 3000) for quantitative analysis. The chromatographic column used was purchased from ThermoFly (Acclaim 300 C18 LC ColuMns, 4.6 mm diameter, 150 mm length, Catalog #: 060266).
A similar method was used to measure the stability of LLC2B-Mal-DM1 and LLC2B-SS-DM1 at room temperature. The peptide was placed at room temperature without plasma incubation, and samples were taken at 0, 30, 60, 120, 240, and 2880 min and used for liquid chromatography mass spectrometry (LC-MS) (Agilent 6545 LC/Q-TOF MS) for quantitative analysis.
2.10. Clone Formation Experiment
We seeded 200 cells per well in 6-well plates with culture medium. After 12 h, the cells were incubated with different agents for 1 week at 37 °C with 5% CO2. Then, we washed the wells with PBS, fixed the cells with methanol for 10 min and stained them with crystal violet at room temperature. Finally, we washed them with sterile water several times in order to obtain a clean background, and then, the colonies formed were photographed and statistically analyzed.
2.11. Tumor-Targeting Analysis via Flow Cytometry
FGFR2-positive cells (80–90%) were dissociated with 0.05% trypsin–EDTA, which was then neutralized with culture medium. To analyze the binding ability of the peptides, the cells (3 × 105) in each sample were incubated with a 2 µL equal amount of LLC2B-Cy5.5 or S-LLC2B-Cy5.5 for 30 min on ice. Then, each sample was washed three times with 1 mL of PBS. Finally, these samples in PBS were analyzed by flow cytometry, and the mean fluorescence intensity (MFI) of each individual sample was evaluated by flow cytometry.
2.12. Antitumor Study In Vivo
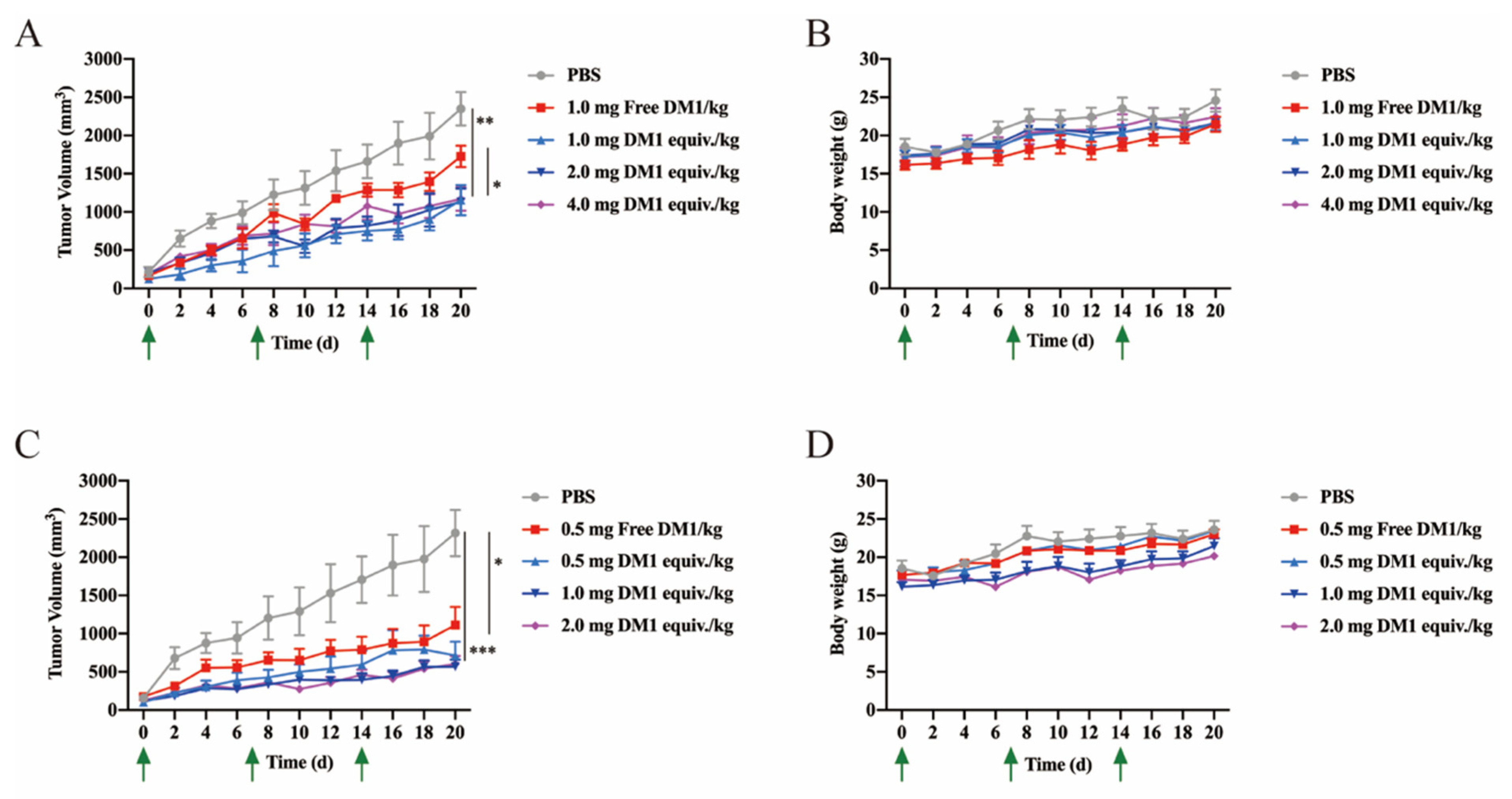
Six-week-old female BALB/c nude mice were purchased from the Animal Experiment Center of Sun Yat-Sen University. A total of 7.0 × 106 MCF-7 cells in 100 μL of PBS were subcutaneously injected into the right flanks of the mice. The tumors were allowed to grow up to 0.3 cm3 (volume = (length × width2)/2). The mice were divided into 9 groups (n = 5 mice/group): PBS, free DM1 (0.5 mg/kg), free DM1 (1.0 mg/kg), LLC2B-Mal-DM1 (1.0 mg DM1 equiv./kg), LLC2B-Mal-DM1 (2.0 mg DM1 equiv./kg), LLC2B-Mal-DM1 (4.0 mg DM1 equiv./kg), LLC2B-SS-DM1 (0.5 mg DM1 equiv./kg), LLC2B-SS-DM1 (1.0 mg DM1 equiv./kg) and LLC2B-SS-DM1 (2.0 mg DM1 equiv./kg). Each mouse was intravenously injected with solutions once a week for 3 weeks, and the tumor volume was measured once every three days for 4 weeks.
2.13. Statistics
All the numerical data are expressed as the mean ± S.D. Statistical differences between two groups were determined by Student’s t test. p < 0.05 was considered significant, (*) for p < 0.05, (**) for p < 0.01 and (***) for p < 0.001.
4. Discussion
The OBOC peptide library used in this study was designed on the basis of the core area of QAE for one short peptide that can bind to FGFR2 found by our team. For the OBOC library constructed on a specific motif, it is easier to obtain desired peptides of high affinity for the target protein. For example, Lam and colleagues designed an OBOC library based on the RGD motif, and screened and obtained peptides named LXW7 and LXW64 for αVβ3 integrins with high specificity. In our study, the selected LLC2B could specifically bind the FGFR2 protein (
Figure 1A). It also had a high binding affinity for FGFR2-positive cancer cells in vitro and in vivo (
Figure 2 and
Figure 3), which indicates the effectiveness of this specific peptide library for screening.
In this study, we also investigated the stability of LLC2B. The results show that it was very stable not only in sterile water but also in plasma. After 144 h, it retained over 90% sequence integrity (
Figure 4). However, in the in vivo assays, we observed that, 24 h after tail vein injection, LLC2B was found to accumulate in the kidneys, except for in tumor tissue in the test group. We concluded that LLC2B had been gradually discharged through metabolic circulation by the body’s urinary system since LLC2B has a low molecular weight. Some of the LLC2B specifically targeted the receptors on the membranes of the tumor cells. Thus, we could observe some extra LLC2B remaining in the kidney after 24 h. At the same time, because LLC2B contains unnatural amino acids, the protease could not recognize and degrade it quickly. The feature also ensures the efficiency of LLC2B in identifying and targeting tumor cells in vivo. It has shown its potential in tumor diagnosis and targeted drug delivery.
Maytansine and its derivatives (maytansinoids DM1 and DM4) are cytotoxins structurally similar to geldanamycin, rifamycin and ansatrienin. They are highly potent inhibitors of microtubule assembly for mitotic arrest and kill tumor cells at subnanomolar concentrations [
23,
24,
25,
26]. However, maytansine failed to be approved as an antitumor agent after clinical trials due to its lack of tumor specificity and unacceptable systemic toxicity [
27]. However, in 2013, an FDA-approved ADC named Kadcyla used DM1 as a cytotoxin to conjugate to the antibody trastuzumab for the treatment of HER2-positive breast cancer. Because of Kadcyla’s good clinical results, we used DM1 as a toxin to similarly construct a PDC in our study. As expected, lower toxicity of DM1 was observed upon conjugating the target ligand LLC2B. Our results prove that LLC2B-SS-DM1 and LLC2B-Mal-DM1 have suppressive effects on the proliferation of FGFR2-positive cells in vitro and in vivo without noticeable toxicity (
Figure 5 and
Figure 6).
In this study, PDCs formed by coupling LLC2B with the toxin DM1 using different linking molecules (3-(2-pyridyldithio) propionic acid and 3-maleimidopropionic acid) had different tumor-inhibiting effects. Our results show that LLC2B-SS-DM1 was better than LLC2B-Mal-DM1 at inhibiting the proliferation of cancer cells (
Figure 5C,D and
Figure 6A,C). This indicates that linker molecules play an important role in the antitumor activity of peptide conjugates. We assume that, after the linker was coupled to DM1, it had some effects on the molecular structure of DM1, which would affect its cell toxicity. Another assumption is that different linker-conjugated toxins have different stabilities, resulting in different leakage effects of DM1, affecting the antitumor effects of PDCs. Therefore, the selection of an appropriate linker is very important for the application of peptide-conjugated drugs. As the toxicity of PDCs in vivo is a major concern for further clinical development, further safety research is necessary to determine which one is more suitable for clinical research between LLC2B-SS-DM1 and LLC2B-Mal-DM1.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





