
Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease

 ,
, 
 , and
, and
Abstract
1. Introduction
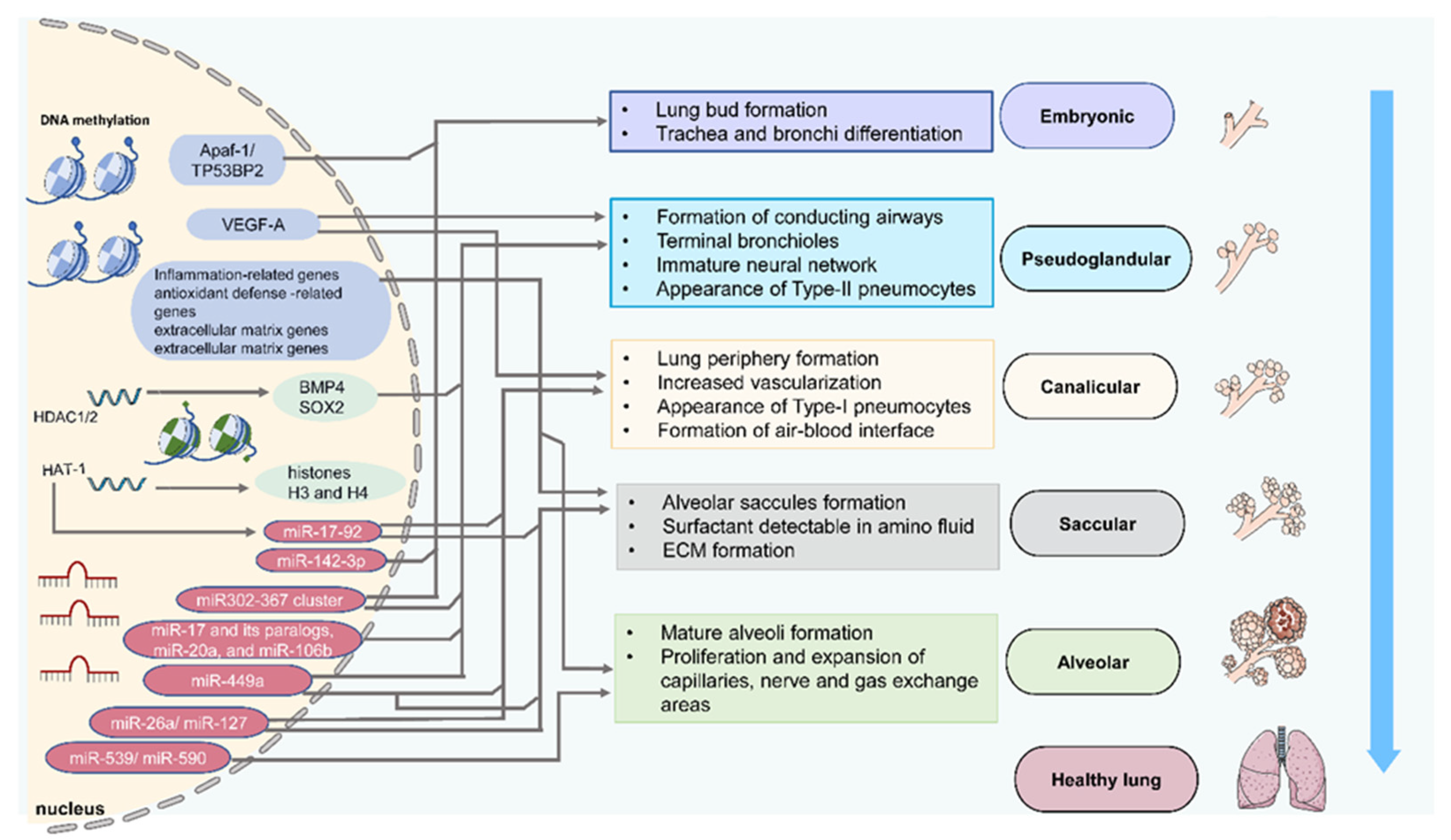
2. Epigenetics and Lung Development
2.1. DNA Methylation
2.2. Histone Acetylation
2.3. MicroRNAs
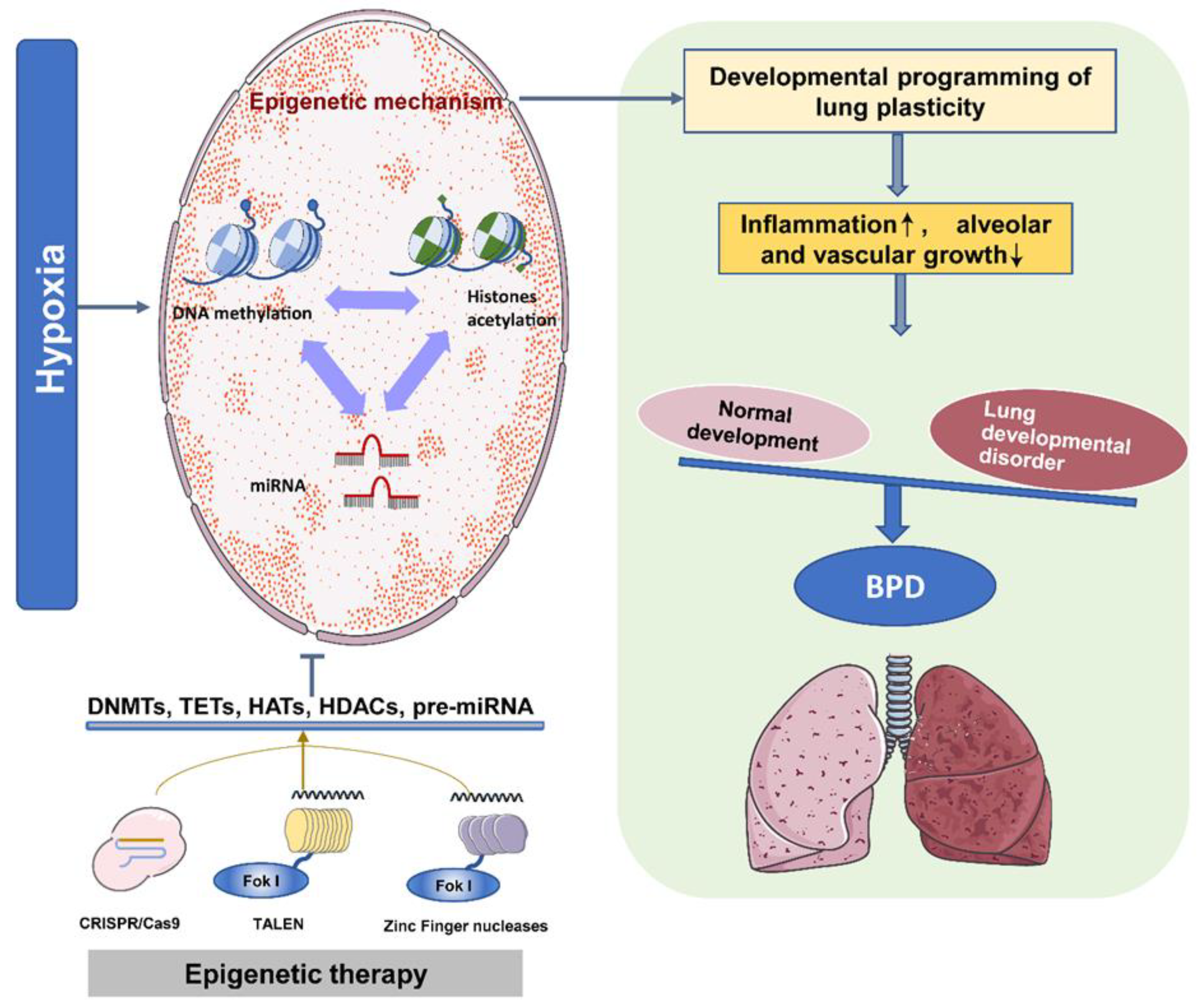
3. Hypoxia and Vulnerability of Neonatal Chronic Lung Disease
4. Epigenetic Programming and Neonatal Chronic Lung Disease

{kind=link}
{kind=link}
{kind=link}
| miRNA | Regulation | Species and Samples | Targets | Disease/Condition | References |
|---|---|---|---|---|---|
| miR-17-92 | Downregulated | Human infant lungs | ? | Extremely and very preterm, BPD | [97] |
| miR-103a-3p and miR-185-5p miR-200a-3p | Downregulated Upregulated | Umbilical cord blood-derived exosomes from human infants | PI3K/Akt and angiogenesis-related signaling pathways | Very preterm, BPD | [98] |
| miR-15a | Upregulated | Chicken lung | Bcl2 | Hypoxia | [99] |
| miR-210 and miR-374a | Upregulated | Plasma of newborn piglets | ? | Hypoxia | [100] |
| miR-34a | Downregulated | Mouse lungs | ? | Postnatal hypoxia-induced BPD | [101] |
5. Perspectives for Treatment and Prevention of Neonatal Chronic Lung Disease
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Moore, L.G.; Charles, S.M.; Julian, C.G. Humans at high altitude: Hypoxia and fetal growth. Respir. Physiol. Neurobiol. 2011, 178, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Amark, H.; Sirotkina, M.; Westgren, M.; Papadogiannakis, N.; Persson, M. Is obesity in pregnancy associated with signs of chronic fetal hypoxia? Acta Obstet. Gynecol. Scand. 2020, 99, 1649–1656. [Google Scholar] [CrossRef]
- Turner, J.M.; Mitchell, M.D.; Kumar, S.S. The physiology of intrapartum fetal compromise at term. Am. J. Obstet. Gynecol. 2020, 222, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhang, L. Epigenetic programming of hypoxic-ischemic encephalopathy in response to fetal hypoxia. Prog. Neurobiol. 2015, 124, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Ducsay, C.A.; Goyal, R.; Pearce, W.J.; Wilson, S.; Hu, X.Q.; Zhang, L. Gestational Hypoxia and Developmental Plasticity. Physiol. Rev. 2018, 98, 1241–1334. [Google Scholar] [CrossRef] [PubMed]
- Gortner, L.; Monz, D.; Mildau, C.; Shen, J.; Kasoha, M.; Laschke, M.W.; Roolfs, T.; Schmiedl, A.; Meier, C.; Tutdibi, E. Bronchopulmonary dysplasia in a double-hit mouse model induced by intrauterine hypoxia and postnatal hyperoxia: Closer to clinical features? Ann. Anat. 2013, 195, 351–358. [Google Scholar] [CrossRef]
- Schmiedl, A.; Roolfs, T.; Tutdibi, E.; Gortner, L.; Monz, D. Influence of prenatal hypoxia and postnatal hyperoxia on morphologic lung maturation in mice. PLoS ONE 2017, 12, e0175804. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mohamed, T.; Abdul-Hafez, A.; Gewolb, I.H.; Uhal, B.D. Oxygen injury in neonates: Which is worse? hyperoxia, hypoxia, or alternating hyperoxia/hypoxia. J. Lung Pulm. Respir. Res. 2020, 7, 4–13. [Google Scholar]
- Valencia, A.M.; Abrantes, M.A.; Hasan, J.; Aranda, J.V.; Beharry, K.D. Reactive Oxygen Species, Biomarkers of Microvascular Maturation and Alveolarization, and Antioxidants in Oxidative Lung Injury. React. Oxyg. Species 2018, 6, 373–388. [Google Scholar] [CrossRef]
- Lorente-Pozo, S.; Parra-Llorca, A.; Lara-Canton, I.; Solaz, A.; Garcia-Jimenez, J.L.; Pallardo, F.V.; Vento, M. Oxygen in the neonatal period: Oxidative stress, oxygen load and epigenetic changes. Semin. Fetal Neonatal Med. 2020, 25, 101090. [Google Scholar] [CrossRef]
- Wang, J.; Dong, W. Oxidative stress and bronchopulmonary dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]
- Narayanan, M.; Owers-Bradley, J.; Beardsmore, C.S.; Mada, M.; Ball, I.; Garipov, R.; Panesar, K.S.; Kuehni, C.E.; Spycher, B.D.; Williams, S.E.; et al. Alveolarization continues during childhood and adolescence: New evidence from helium-3 magnetic resonance. Am. J. Respir. Crit. Care Med. 2012, 185, 186–191. [Google Scholar] [CrossRef]
- Thekkeveedu, R.K.; Guaman, M.C.; Shivanna, B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med. 2017, 132, 170–177. [Google Scholar] [CrossRef]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef]
- Kramer, B.W.; Lievense, S.; Been, J.V.; Zimmermann, L.J. From classic to new bronchopulmonary dysplasia. Ned. Tijdschr. Geneeskd. 2010, 154, A1024. [Google Scholar]
- Baker, C.D.; Alvira, C.M. Disrupted lung development and bronchopulmonary dysplasia: Opportunities for lung repair and regeneration. Curr. Opin. Pediatr. 2014, 26, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A. “New” bronchopulmonary dysplasia and chronic lung disease. Paediatr. Respir. Rev. 2017, 24, 17–18. [Google Scholar] [CrossRef]
- Silva, D.M.; Nardiello, C.; Pozarska, A.; Morty, R.E. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1239–L1272. [Google Scholar] [CrossRef]
- Collins, J.J.P.; Tibboel, D.; de Kleer, I.M.; Reiss, I.K.M.; Rottier, R.J. The Future of Bronchopulmonary Dysplasia: Emerging Pathophysiological Concepts and Potential New Avenues of Treatment. Front. Med. 2017, 4, 61. [Google Scholar] [CrossRef]
- Abman, S.H.; Bancalari, E.; Jobe, A. The Evolution of Bronchopulmonary Dysplasia after 50 Years. Am. J. Respir. Crit. Care Med. 2017, 195, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R. Signaling Pathways Involved in the Development of Bronchopulmonary Dysplasia and Pulmonary Hypertension. Children 2020, 7, 100. [Google Scholar] [CrossRef]
- Nanduri, J.; Makarenko, V.; Reddy, V.D.; Yuan, G.; Pawar, A.; Wang, N.; Khan, S.A.; Zhang, X.; Kinsman, B.; Peng, Y.J.; et al. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 2515–2520. [Google Scholar] [CrossRef]
- Palma-Gudiel, H.; Eixarch, E.; Crispi, F.; Moran, S.; Zannas, A.S.; Fananas, L. Prenatal adverse environment is associated with epigenetic age deceleration at birth and hypomethylation at the hypoxia-responsive EP300 gene. Clin. Epigenet. 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Pasha, A.B.; Chen, X.Q.; Zhou, G.P. Bronchopulmonary dysplasia: Pathogenesis and treatment. Exp. Ther. Med. 2018, 16, 4315–4321. [Google Scholar] [CrossRef]
- Rood, K.; Lopez, V.; La Frano, M.R.; Fiehn, O.; Zhang, L.; Blood, A.B.; Wilson, S.M. Gestational Hypoxia and Programing of Lung Metabolism. Front. Physiol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetic control of gene expression in the lung. Am. J. Respir. Crit. Care Med. 2011, 183, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Hagood, J.S. Beyond the genome: Epigenetic mechanisms in lung remodeling. Physiology 2014, 29, 177–185. [Google Scholar] [CrossRef]
- Aboud, N.M.A.; Shahid, H.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hoareau-Aveilla, C.; Meggetto, F. Crosstalk between microRNA and DNA Methylation Offers Potential Biomarkers and Targeted Therapies in ALK-Positive Lymphomas. Cancers 2017, 9, 100. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Wang, X.; Liu, Y.; Gu, B.; Zhao, G.; Li, Y. Methylation of TP53BP2 and Apaf-1 genes in embryonic lung cells and their impact on gene expression. Ann. Transl. Med. 2018, 6, 459. [Google Scholar] [CrossRef]
- Nagarajan, P.; Ge, Z.; Sirbu, B.; Doughty, C.; Garcia, P.A.A.; Schlederer, M.; Annunziato, A.T.; Cortez, D.; Kenner, L.; Parthun, M.R. Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet. 2013, 9, e1003518. [Google Scholar] [CrossRef]
- Carraro, G.; Shrestha, A.; Rostkovius, J.; Contreras, A.; Chao, C.M.; El Agha, E.; Mackenzie, B.; Dilai, S.; Guidolin, D.; Taketo, M.M.; et al. miR-142-3p balances proliferation and differentiation of mesenchymal cells during lung development. Development 2014, 141, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Cushing, L.; Ai, X.; Lu, J. miR-326 is downstream of Sonic hedgehog signaling and regulates the expression of Gli2 and smoothened. Am. J. Respir. Cell Mol. Biol. 2014, 51, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.J.; Land, S.C. Regulation of vascular signalling by nuclear Sprouty2 in fetal lung epithelial cells: Implications for co-ordinated airway and vascular branching in lung development. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2018, 224, 105–114. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, Y.; Morley, M.P.; Lu, M.M.; Demayo, F.J.; Olson, E.N.; Morrisey, E.E. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 2013, 24, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Carraro, G.; El-Hashash, A.; Guidolin, D.; Tiozzo, C.; Turcatel, G.; Young, B.M.; De Langhe, S.P.; Bellusci, S.; Shi, W.; Parnigotto, P.P.; et al. miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev. Biol. 2009, 333, 238–250. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Y.; Hurd, L.; Hannenhalli, S.; Liu, F.; Lu, M.M.; Morrisey, E.E. Regulation of lung endoderm progenitor cell behavior by miR302/367. Development 2011, 138, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Sanford, E.L.; Choy, K.W.; Donahoe, P.K.; Tracy, A.A.; Hila, R.; Loscertales, M.; Longoni, M. MiR-449a Affects Epithelial Proliferation during the Pseudoglandular and Canalicular Phases of Avian and Mammal Lung Development. PLoS ONE 2016, 11, e0149425. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Wang, Y.; Zhang, H.; Weng, T.; Baviskar, P.; Guo, Y.; Gou, D.; Liu, L. MicroRNA-127 modulates fetal lung development. Physiol. Genom. 2009, 37, 268–278. [Google Scholar] [CrossRef]
- Sun, Y.F.; Kan, Q.; Yang, Y.; Zhang, Y.H.; Shen, J.X.; Zhang, C.; Zhou, X.Y. Knockout of microRNA26a promotes lung development and pulmonary surfactant synthesis. Mol. Med. Rep. 2018, 17, 5988–5995. [Google Scholar] [CrossRef]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef]
- Cuna, A.; Halloran, B.; Faye-Petersen, O.; Kelly, D.; Crossman, D.K.; Cui, X.; Pandit, K.; Kaminski, N.; Bhattacharya, S.; Ahmad, A.; et al. Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am. J. Respir. Cell Mol. Biol. 2015, 53, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ahangari, F.; Espinoza, C.R.; Chhabra, D.; Nicola, T.; Yan, X.; Lal, C.V.; Hagood, J.S.; Kaminski, N.; Bar-Joseph, Z.; et al. Integrating multiomics longitudinal data to reconstruct networks underlying lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L556–L568. [Google Scholar] [CrossRef]
- Kumar, S.; Chinnusamy, V.; Mohapatra, T. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Front. Genet. 2018, 9, 640. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. Basic science: Epigenetic programming and the respiratory system. Breathe 2013, 9, 278–288. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef]
- Fehl, J.; Pozarska, A.; Nardiello, C.; Rath, P.; Solaligue, D.E.S.; Vadasz, I.; Mayer, K.; Herold, S.; Seeger, W.; Morty, R.E. Control Interventions Can Impact Alveolarization and the Transcriptome in Developing Mouse Lungs. Anat. Rec. 2019, 302, 346–363. [Google Scholar] [CrossRef]
- Merid, S.K.; Novoloaca, A.; Sharp, G.C.; Kupers, L.K.; Kho, A.T.; Roy, R.; Gao, L.; Annesi-Maesano, I.; Jain, P.; Plusquin, M.; et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med. 2020, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Bemer, M. Unraveling the Complex Epigenetic Mechanisms that Regulate Gene Activity. Methods Mol. Biol. 2018, 1675, 205–231. [Google Scholar] [CrossRef]
- Joss-Moore, L.A.; Albertine, K.H.; Lane, R.H. Epigenetics and the developmental origins of lung disease. Mol. Genet. Metab. 2011, 104, 61–66. [Google Scholar] [CrossRef]
- Khan, S.N.; Khan, A.U. Role of histone acetylation in cell physiology and diseases: An update. Clin. Chim. Acta 2010, 411, 1401–1411. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Alisch, R.S.; Jin, P.; Epstein, M.; Caspary, T.; Warren, S.T. Argonaute2 is essential for mammalian gastrulation and proper mesoderm formation. PLoS Genet. 2007, 3, e227. [Google Scholar] [CrossRef]
- Muller, M.; Fazi, F.; Ciaudo, C. Argonaute Proteins: From Structure to Function in Development and Pathological Cell Fate Determination. Front. Cell Dev. Biol. 2019, 7, 360. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Zhang, Z.; McManus, M.T.; Harfe, B.D.; Sun, X. Dicer function is essential for lung epithelium morphogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 2208–2213. [Google Scholar] [CrossRef]
- Yang, Y.; Kai, G.; Pu, X.D.; Qing, K.; Guo, X.R.; Zhou, X.Y. Expression profile of microRNAs in fetal lung development of Sprague-Dawley rats. Int. J. Mol. Med. 2012, 29, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Ye, X.Y.; Singhal, N.; De La Rue, S.; Lodha, A.; Shah, P.S. Higher altitude and risk of bronchopulmonary dysplasia among preterm infants. Am. J. Perinatol. 2013, 30, 601–606. [Google Scholar] [CrossRef]
- Alshehri, M.A. Are preterm infants at high altitude at greater risk for the development of bronchopulmonary dysplasia? J. Trop. Pediatr. 2014, 60, 68–73. [Google Scholar] [CrossRef]
- Hu, X.Q.; Huang, X.; Xiao, D.; Zhang, L. Direct effect of chronic hypoxia in suppressing large conductance Ca(2+)-activated K(+) channel activity in ovine uterine arteries via increasing oxidative stress. J. Physiol. 2016, 594, 343–356. [Google Scholar] [CrossRef]
- Xiao, D.; Hu, X.Q.; Huang, X.; Zhou, J.; Wilson, S.M.; Yang, S.; Zhang, L. Chronic hypoxia during gestation enhances uterine arterial myogenic tone via heightened oxidative stress. PLoS ONE 2013, 8, e73731. [Google Scholar] [CrossRef]
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444. [Google Scholar] [CrossRef]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef]
- Tracy, M.K.; Berkelhamer, S.K. Bronchopulmonary Dysplasia and Pulmonary Outcomes of Prematurity. Pediatr. Ann. 2019, 48, e148–e153. [Google Scholar] [CrossRef]
- Hislop, A. Developmental biology of the pulmonary circulation. Paediatr. Respir. Rev. 2005, 6, 35–43. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular endothelial growth factor in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Alvira, C.M. Aberrant Pulmonary Vascular Growth and Remodeling in Bronchopulmonary Dysplasia. Front. Med. 2016, 3, 21. [Google Scholar] [CrossRef]
- Hocq, C.; Vanhoutte, L.; Guilloteau, A.; Massolo, A.C.; Van Grambezen, B.; Carkeek, K.; Piersigilli, F.; Danhaive, O.; from the European Society for Pediatric Research. Early diagnosis and targeted approaches to pulmonary vascular disease in bronchopulmonary dysplasia. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Mandell, E.W.; Abman, S.H. Fetal Vascular Origins of Bronchopulmonary Dysplasia. J. Pediatr. 2017, 185, 7–10. [Google Scholar] [CrossRef]
- Remesal, A.; Pedraz, C.; San Feliciano, L.; Ludena, D. Pulmonary expression of vascular endothelial growth factor (VEGF) and alveolar septation in a newborn rat model exposed to acute hypoxia and recovered under conditions of air or hyperoxia. Histol. Histopathol. 2009, 24, 325–330. [Google Scholar] [CrossRef]
- Remesal, A.; San Feliciano, L.; Isidoro-Garcia, M.; Ludena, D. Effects of antenatal betamethasone and dexamethasone on the lung expression of vascular endothelial growth factor and alveolarization in newborn rats exposed to acute hypoxia and recovered in normoxia or hyperoxia. Neonatology 2010, 98, 313–320. [Google Scholar] [CrossRef]
- Gallacher, D.J.; Hart, K.; Kotecha, S. Common respiratory conditions of the newborn. Breathe 2016, 12, 30–42. [Google Scholar] [CrossRef]
- Filby, C.E.; Hooper, S.B.; Wallace, M.J. Partial pulmonary embolization disrupts alveolarization in fetal sheep. Respir. Res. 2010, 11, 42. [Google Scholar] [CrossRef]
- Kang, N.Y.; Ivanovska, J.; Tamir-Hostovsky, L.; Belik, J.; Gauda, E.B. Chronic Intermittent Hypoxia in Premature Infants: The Link Between Low Fat Stores, Adiponectin Receptor Signaling and Lung Injury. Adv. Exp. Med. Biol. 2018, 1071, 151–157. [Google Scholar] [CrossRef]
- Schittny, J.C.; Mund, S.I.; Stampanoni, M. Evidence and structural mechanism for late lung alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L246–L254. [Google Scholar] [CrossRef]
- Mund, S.I.; Stampanoni, M.; Schittny, J.C. Developmental alveolarization of the mouse lung. Dev. Dyn. 2008, 237, 2108–2116. [Google Scholar] [CrossRef]
- Elberson, V.D.; Nielsen, L.C.; Wang, H.; Kumar, H.S. Effects of intermittent hypoxia and hyperoxia on angiogenesis and lung development in newborn mice. J. Neonatal Perinat. Med. 2015, 8, 313–322. [Google Scholar] [CrossRef]
- Chao, C.M.; Moiseenko, A.; Zimmer, K.P.; Bellusci, S. Alveologenesis: Key cellular players and fibroblast growth factor 10 signaling. Mol. Cell. Pediatr. 2016, 3, 17. [Google Scholar] [CrossRef]
- Rodriguez-Castillo, J.A.; Perez, D.B.; Ntokou, A.; Seeger, W.; Morty, R.E.; Ahlbrecht, K. Understanding alveolarization to induce lung regeneration. Respir. Res. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Mammoto, T. Vascular Niche in Lung Alveolar Development, Homeostasis, and Regeneration. Front. Bioeng. Biotechnol. 2019, 7, 318. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Taylor, D.; Ovchinnikov, D.A.; Wolvetang, E.J.; de Torrente, L.; Mar, J.C. Variability of Gene Expression Identifies Transcriptional Regulators of Early Human Embryonic Development. PLoS Genet. 2015, 11, e1005428. [Google Scholar] [CrossRef]
- Dumeige, L.; Nehlich, M.; Viengchareun, S.; Perrot, J.; Pussard, E.; Lombes, M.; Martinerie, L. Preterm birth is associated with epigenetic programming of transgenerational hypertension in mice. Exp. Mol. Med. 2020, 52, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Vogel, E.R.; Britt, R.D., Jr.; Trinidad, M.C.; Faksh, A.; Martin, R.J.; MacFarlane, P.M.; Pabelick, C.M.; Prakash, Y.S. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 2015, 93, 119–127. [Google Scholar] [CrossRef]
- Lafuente, E.; Beldade, P. Genomics of Developmental Plasticity in Animals. Front. Genet. 2019, 10, 720. [Google Scholar] [CrossRef]
- Meiners, S.; Hilgendorff, A. Early injury of the neonatal lung contributes to premature lung aging: A hypothesis. Mol. Cell. Pediatr. 2016, 3, 24. [Google Scholar] [CrossRef]
- McEvoy, C.T.; Spindel, E.R. Pulmonary Effects of Maternal Smoking on the Fetus and Child: Effects on Lung Development, Respiratory Morbidities, and Life Long Lung Health. Paediatr. Respir. Rev. 2017, 21, 27–33. [Google Scholar] [CrossRef]
- Lignelli, E.; Palumbo, F.; Myti, D.; Morty, R.E. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L832–L887. [Google Scholar] [CrossRef]
- Sahoo, D.; Zaramela, L.S.; Hernandez, G.E.; Mai, U.; Taheri, S.; Dang, D.; Stouch, A.N.; Medal, R.M.; McCoy, A.M.; Aschner, J.L.; et al. Transcriptional profiling of lung macrophages identifies a predictive signature for inflammatory lung disease in preterm infants. Commun. Biol. 2020, 3, 259. [Google Scholar] [CrossRef]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. J. Clin. Investig. Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Chen, W.J.; Hsieh, W.S.; Tsao, P.N.; Yu, S.L.; Lai, C.Y.; Lee, W.C.; Jeng, S.F. MicroRNA expression aberration associated with bronchopulmonary dysplasia in preterm infants: A preliminary study. Respir. Care 2013, 58, 1527–1535. [Google Scholar] [CrossRef]
- Wong, M.J.; Kantores, C.; Ivanovska, J.; Jain, A.; Jankov, R.P. Simvastatin prevents and reverses chronic pulmonary hypertension in newborn rats via pleiotropic inhibition of RhoA signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L985–L999. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, A.; Alphonse, R.S.; Collins, J.J.; van Haaften, T.; O’Reilly, M.; Eaton, F.; Thebaud, B. The axonal guidance cue semaphorin 3C contributes to alveolar growth and repair. PLoS ONE 2013, 8, e67225. [Google Scholar] [CrossRef]
- Ahmad, A.; Bhattacharya, S.; Sridhar, A.; Iqbal, A.M.; Mariani, T.J. Recurrent copy number variants associated with bronchopulmonary dysplasia. Pediatr. Res. 2016, 79, 940–945. [Google Scholar] [CrossRef][Green Version]
- Rogers, L.K.; Robbins, M.; Dakhlallah, D.; Yang, Z.; Lee, L.J.; Mikhail, M.; Nuovo, G.; Pryhuber, G.S.; McGwin, G.; Marsh, C.B.; et al. Attenuation of miR-17 approximately 92 Cluster in Bronchopulmonary Dysplasia. Ann. Am. Thorac. Soc. 2015, 12, 1506–1513. [Google Scholar] [CrossRef]
- Zhong, X.Q.; Yan, Q.; Chen, Z.G.; Jia, C.H.; Li, X.H.; Liang, Z.Y.; Gu, J.; Wei, H.L.; Lian, C.Y.; Zheng, J.; et al. Umbilical Cord Blood-Derived Exosomes From Very Preterm Infants With Bronchopulmonary Dysplasia Impaired Endothelial Angiogenesis: Roles of Exosomal MicroRNAs. Front. Cell Dev. Biol. 2021, 9, 637248. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Hu, X.; Wu, C.; Li, N. Hypoxia-induced miR-15a promotes mesenchymal ablation and adaptation to hypoxia during lung development in chicken. PLoS ONE 2014, 9, e98868. [Google Scholar] [CrossRef]
- Garberg, H.T.; Huun, M.U.; Baumbusch, L.O.; Asegg-Atneosen, M.; Solberg, R.; Saugstad, O.D. Temporal Profile of Circulating microRNAs after Global Hypoxia-Ischemia in Newborn Piglets. Neonatology 2017, 111, 133–139. [Google Scholar] [CrossRef]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- MacDonald, V.E.; Howe, L.J. Histone acetylation: Where to go and how to get there. Epigenetics 2009, 4, 139–143. [Google Scholar] [CrossRef]
- Engelen, E.; Brandsma, J.H.; Moen, M.J.; Signorile, L.; Dekkers, D.H.; Demmers, J.; Kockx, C.E.; Ozgur, Z.; van IJcken, W.F.J.; van den Berg, D.L.C.; et al. Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat. Commun. 2015, 6, 7155. [Google Scholar] [CrossRef]
- Barnes, P.J.; Adcock, I.M.; Ito, K. Histone acetylation and deacetylation: Importance in inflammatory lung diseases. Eur. Respir. J. 2005, 25, 552–563. [Google Scholar] [CrossRef]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Ma, X.L.; Shen, Z.; Wu, X.L.; Cheng, F.; Du, L.Z. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 2010, 28, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Masjedi, M.R.; Barnes, P.J.; Adcock, I.M. Epigenetics and chromatin remodeling play a role in lung disease. Tanaffos 2011, 10, 7–16. [Google Scholar]
- Yao, Y.; Liu, Q.; Adrianto, I.; Wu, X.; Glassbrook, J.; Khalasawi, N.; Yin, C.; Yi, Q.; Dong, Z.; Geissmann, F.; et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020, 11, 3822. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Circ. 2013, 3, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Rahman, M.; Nana-Sinkam, S.P. MicroRNAs in respiratory disease. A clinician’s overview. Ann. Am. Thorac. Soc. 2014, 11, 1277–1285. [Google Scholar] [CrossRef]
- Nardiello, C.; Morty, R.E. MicroRNA in late lung development and bronchopulmonary dysplasia: The need to demonstrate causality. Mol. Cell. Pediatr. 2016, 3, 19. [Google Scholar] [CrossRef]
- Olave, N.; Lal, C.V.; Halloran, B.; Pandit, K.; Cuna, A.C.; Faye-Petersen, O.M.; Kelly, D.R.; Nicola, T.; Benos, P.V.; Kaminski, N.; et al. Regulation of alveolar septation by microRNA-489. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L476–L487. [Google Scholar] [CrossRef]
- Herrera-Rivero, M.; Zhang, R.; Heilmann-Heimbach, S.; Mueller, A.; Bagci, S.; Dresbach, T.; Schroder, L.; Holdenrieder, S.; Reutter, H.M.; Kipfmueller, F. Circulating microRNAs are associated with Pulmonary Hypertension and Development of Chronic Lung Disease in Congenital Diaphragmatic Hernia. Sci. Rep. 2018, 8, 10735. [Google Scholar] [CrossRef]
- Dutta, R.K.; Chinnapaiyan, S.; Unwalla, H. Aberrant MicroRNAomics in Pulmonary Complications: Implications in Lung Health and Diseases. Mol. Ther. Nucleic Acids 2019, 18, 413–431. [Google Scholar] [CrossRef]
- White, K.; Lu, Y.; Annis, S.; Hale, A.E.; Chau, B.N.; Dahlman, J.E.; Hemann, C.; Opotowsky, A.R.; Vargas, S.O.; Rosas, I.; et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 2015, 7, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; MacIntyre, D.A.; Binkhamis, R.; Cook, J.; Sykes, L.; Bennett, P.R.; Terzidou, V. Maternal plasma miRNAs as potential biomarkers for detecting risk of small-for-gestational-age births. EBioMedicine 2020, 62, 103145. [Google Scholar] [CrossRef]
- Patterson, A.J.; Xiao, D.; Xiong, F.; Dixon, B.; Zhang, L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovasc. Res. 2012, 93, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, M.; Barkhuizen, M.; van den Hove, D.L.A.; Steinbusch, H.W.M.; Bruno, M.A.; Loidl, C.F.; Gavilanes, A.W.D. Clinical Implications of Epigenetic Dysregulation in Perinatal Hypoxic-Ischemic Brain Damage. Front. Neurol. 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Yu, R.M.K.; Wu, R.S.S.; Kong, R.Y.C. Overexpression and Knockdown of Hypoxia-Inducible Factor 1 Disrupt the Expression of Steroidogenic Enzyme Genes and Early Embryonic Development in Zebrafish. Gene Regul. Syst. Biol. 2017, 11. [Google Scholar] [CrossRef]
- Pamenter, M.E.; Hall, J.E.; Tanabe, Y.; Simonson, T.S. Cross-Species Insights Into Genomic Adaptations to Hypoxia. Front. Genet. 2020, 11, 743. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Tyson, J.E.; Wright, L.L.; Oh, W.; Kennedy, K.A.; Mele, L.; Ehrenkranz, R.A.; Stoll, B.J.; Lemons, J.A.; Stevenson, D.K.; Bauer, C.R.; et al. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N. Engl. J. Med. 1999, 340, 1962–1968. [Google Scholar] [CrossRef] [PubMed]
- Londhe, V.A.; Nolen, T.L.; Das, A.; Higgins, R.D.; Tyson, J.E.; Oh, W.; Devaskar, S.U. Vitamin A supplementation in extremely low-birth-weight infants: Subgroup analysis in small-for-gestational-age infants. Am. J. Perinatol. 2013, 30, 771–780. [Google Scholar] [CrossRef]
- Pakvasa, M.A.; Saroha, V.; Patel, R.M. Optimizing Caffeine Use and Risk of Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review, Meta-analysis, and Application of Grading of Recommendations Assessment, Development, and Evaluation Methodology. Clin. Perinatol. 2018, 45, 273–291. [Google Scholar] [CrossRef]
- Baud, O.; Watterberg, K.L. Prophylactic postnatal corticosteroids: Early hydrocortisone. Semin. Fetal Neonatal Med. 2019, 24, 202–206. [Google Scholar] [CrossRef]
- Cetinkaya, M.; Cansev, M.; Cekmez, F.; Tayman, C.; Canpolat, F.E.; Kafa, I.M.; Yaylagul, E.O.; Kramer, B.W.; Sarici, S.U. Protective Effects of Valproic Acid, a Histone Deacetylase Inhibitor, against Hyperoxic Lung Injury in a Neonatal Rat Model. PLoS ONE 2015, 10, e0126028. [Google Scholar] [CrossRef] [PubMed]
- Menden, H.; Xia, S.; Mabry, S.M.; Noel-MacDonnell, J.; Rajasingh, J.; Ye, S.Q.; Sampath, V. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L332–L346. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, L.; Yu, J.; Fu, H.; Zhou, D.; Liu, H. Inhibition of microRNA-29a alleviates hyperoxia-induced bronchopulmonary dysplasia in neonatal mice via upregulation of GAB1. Mol. Med. 2019, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hong, H.; Li, X.-X.; Li, J.; Zhang, Z.-Q. The involvement of HDAC3-mediated inhibition of microRNA cluster 17-92 in hyperoxia-mediated impairment of lung development in neonatal rats. bioRxiv 2020, 1–26. [Google Scholar] [CrossRef]
- David, A.L.; Waddington, S.N. Candidate diseases for prenatal gene therapy. Methods Mol. Biol. 2012, 891, 9–39. [Google Scholar] [CrossRef]
- Cathomen, T.; Joung, J.K. Zinc-finger nucleases: The next generation emerges. Mol. Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Scott, A. How CRISPR is transforming drug discovery. Nature 2018, 555, S10–S11. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Tong, R.; Li, M.; Liu, Y.; Xue, J.; Lu, Y. Advancements and Obstacles of CRISPR-Cas9 Technology in Translational Research. Mol. Ther. Methods Clin. Dev. 2019, 13, 359–370. [Google Scholar] [CrossRef]
- Schacker, M.; Seimetz, D. From fiction to science: Clinical potentials and regulatory considerations of gene editing. Clin. Transl. Med. 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Lu, A.; Wang, J.; Sun, W.; Huang, W.; Cai, Z.; Zhao, G.; Wang, J. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019, 5, 22. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef]
- Sapozhnikov, D.M.; Szyf, M. Unraveling the functional role of DNA methylation using targeted DNA demethylation by steric blockage of DNA methyltransferase with CRISPR/dCas9. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Chen, L.F.; Lin, Y.T.; Gallegos, D.A.; Hazlett, M.F.; Gomez-Schiavon, M.; Yang, M.G.; Kalmeta, B.; Zhou, A.S.; Holtzman, L.; Gersbach, C.A.; et al. Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep. 2019, 26, 1174–1188. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, X.; Wei, M.; Gao, X.; Zhao, L.; Shi, R.; Sun, W.; Duan, Y.; Yang, G.; Yuan, L. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, K.; Cheng, Y.; Zhong, Z.; Tian, L.; Tang, X.; Tang, A.; Zheng, X.; Zhang, T.; Qi, Y.; et al. CRISPR-Cas9 Based Genome Editing Reveals New Insights into MicroRNA Function and Regulation in Rice. Front. Plant Sci. 2017, 8, 1598. [Google Scholar] [CrossRef]
- Basso, M.F.; Ferreira, P.C.G.; Kobayashi, A.K.; Harmon, F.G.; Nepomuceno, A.L.; Molinari, H.B.C.; Grossi-de-Sa, M.F. MicroRNAs and new biotechnological tools for its modulation and improving stress tolerance in plants. Plant Biotechnol. J. 2019, 17, 1482–1500. [Google Scholar] [CrossRef]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; Lopez-Giraldez, F.; Coskun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Alapati, D.; Morrisey, E.E. Gene Editing and Genetic Lung Disease. Basic Research Meets Therapeutic Application. Am. J. Respir. Cell Mol. Biol. 2017, 56, 283–290. [Google Scholar] [CrossRef]
- Hodges, C.A.; Conlon, R.A. Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 2019, 6, 97–108. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.H.; Lee, M.S.; Kim, M.S. Epigenetic modification by dietary factors: Implications in metabolic syndrome. Mol. Aspects Med. 2017, 54, 58–70. [Google Scholar] [CrossRef]
- Li, S.; Chen, M.; Li, Y.; Tollefsbol, T.O. Prenatal epigenetics diets play protective roles against environmental pollution. Clin. Epigenet. 2019, 11, 82. [Google Scholar] [CrossRef]
- Mao, X.; Qiu, J.; Zhao, L.; Xu, J.; Yin, J.; Yang, Y.; Zhang, M.; Cheng, R. Vitamin D and IL-10 Deficiency in Preterm Neonates With Bronchopulmonary Dysplasia. Front. Pediatr. 2018, 6, 246. [Google Scholar] [CrossRef]
- Park, H.W.; Lim, G.; Park, Y.M.; Chang, M.; Son, J.S.; Lee, R. Association between vitamin D level and bronchopulmonary dysplasia: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0235332. [Google Scholar] [CrossRef]
- Mandell, E.; Seedorf, G.; Gien, J.; Abman, S.H. Vitamin D treatment improves survival and infant lung structure after intra-amniotic endotoxin exposure in rats: Potential role for the prevention of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L420–L428. [Google Scholar] [CrossRef]
- Morty, R.E. Using Experimental Models to Identify Pathogenic Pathways and Putative Disease Management Targets in Bronchopulmonary Dysplasia. Neonatology 2020, 117, 233–239. [Google Scholar] [CrossRef]



| Stage | Species and Samples | Epigenetics | Targeted Genes | Characteristic Events | References |
|---|---|---|---|---|---|
| Embryonic | Human embryonic lung cells | DNA Methylation | TP53BP2 and Apaf-1 | Cell proliferation | [31] |
| Mouse embryonic fibroblasts | HAT-1 | Histones H3 and H4 | Embryonic lung development | [32] | |
| Mouse lung primordia | miR-142-3p | WNT signaling | Lung mesenchymal cells proliferation and differentiation | [33] | |
| Mouse embryonic lung explants | miR-326 | Smo and Gli2 | Lung mesenchymal cells proliferation and differentiation | [34] | |
| Pseudoglandular | Rat fetal distal lung epithelial cells | DNA Methylation | VEGF-A | Airway and vascular branching | [35] |
| Mouse proximal lung endoderm progenitors | HDAC1/2 | BMP4/SOX2 | Branching morphogenesis | [36] | |
| Mouse embryonic lung epithelial explants | miR-17 and its paralogs, miR-20a, and miR-106b | Stat3 and Mapk14 | Epithelial bud morphogenesis | [37] | |
| Early lung endoderm | miR302–367 cluster | Rbl2 and Cdkn1a | Lung epithelial proliferation | [38] | |
| Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] | |
| Rat fetal lungs | miR-127 | Lung branching | [40] | ||
| Canalicular | Rat fetal distal lung epithelial cells | DNA Methylation | VEGF-A | Airway and vascular branching | [35] |
| Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] | |
| Mouse fetal lungs | miR-26a | SFTPA1, SFTPB, SFTPC | Formation of dilated lumens and aerated regions, maturation of the alveolar structure | [41] | |
| Saccular | Human, murine, and avian fetal lungs | miR-449a | Mycn and Sox9 | Epithelial proliferation and mucociliary differentiation | [39] |
| miR-26a | SFTPA1, SFTPB, SFTPC | Maturation of the alveolar structure | [41] | ||
| Mouse fetal lungs | HDAC3/miR-17-92 | TGF-β | Alveolar type 1 cell spreading and lung sacculation | [42] | |
| Alveolar | Mouse newborn lung | DNA Methylation | ? | Alveolar septation | [43] |
| Mouse postnatal and early child lung | miR-539 and miR-590 | Alveolar development | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, Y.; Zhang, S.; Riddle, S.; Zhang, L.; Song, R.; Yue, D. Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease. Biomedicines 2021, 9, 944. https://doi.org/10.3390/biomedicines9080944
Tong Y, Zhang S, Riddle S, Zhang L, Song R, Yue D. Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease. Biomedicines. 2021; 9(8):944. https://doi.org/10.3390/biomedicines9080944
Chicago/Turabian StyleTong, Yajie, Shuqing Zhang, Suzette Riddle, Lubo Zhang, Rui Song, and Dongmei Yue. 2021. "Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease" Biomedicines 9, no. 8: 944. https://doi.org/10.3390/biomedicines9080944
APA StyleTong, Y., Zhang, S., Riddle, S., Zhang, L., Song, R., & Yue, D. (2021). Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease. Biomedicines, 9(8), 944. https://doi.org/10.3390/biomedicines9080944