
Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration

 ,
, 

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthetic NHK1 Peptides
2.2. Cell Cultures, Mantainance, and Viability
2.3. Membrane Permeability Assay by Transwell System
2.4. Fluorescence Mycroscopy
2.5. High-Resolution Respirometry
2.6. Analysis of Respirometric States
2.7. Cell Lysates and Fractionation
2.8. Western Blot Analysis
2.9. Real-Time PCR
2.10. Flow Cytometry Experiments
2.11. Cell Transfection
2.12. Pull-Down Assay
2.13. Docking Simulation
2.14. Statistical Analysis
3. Results
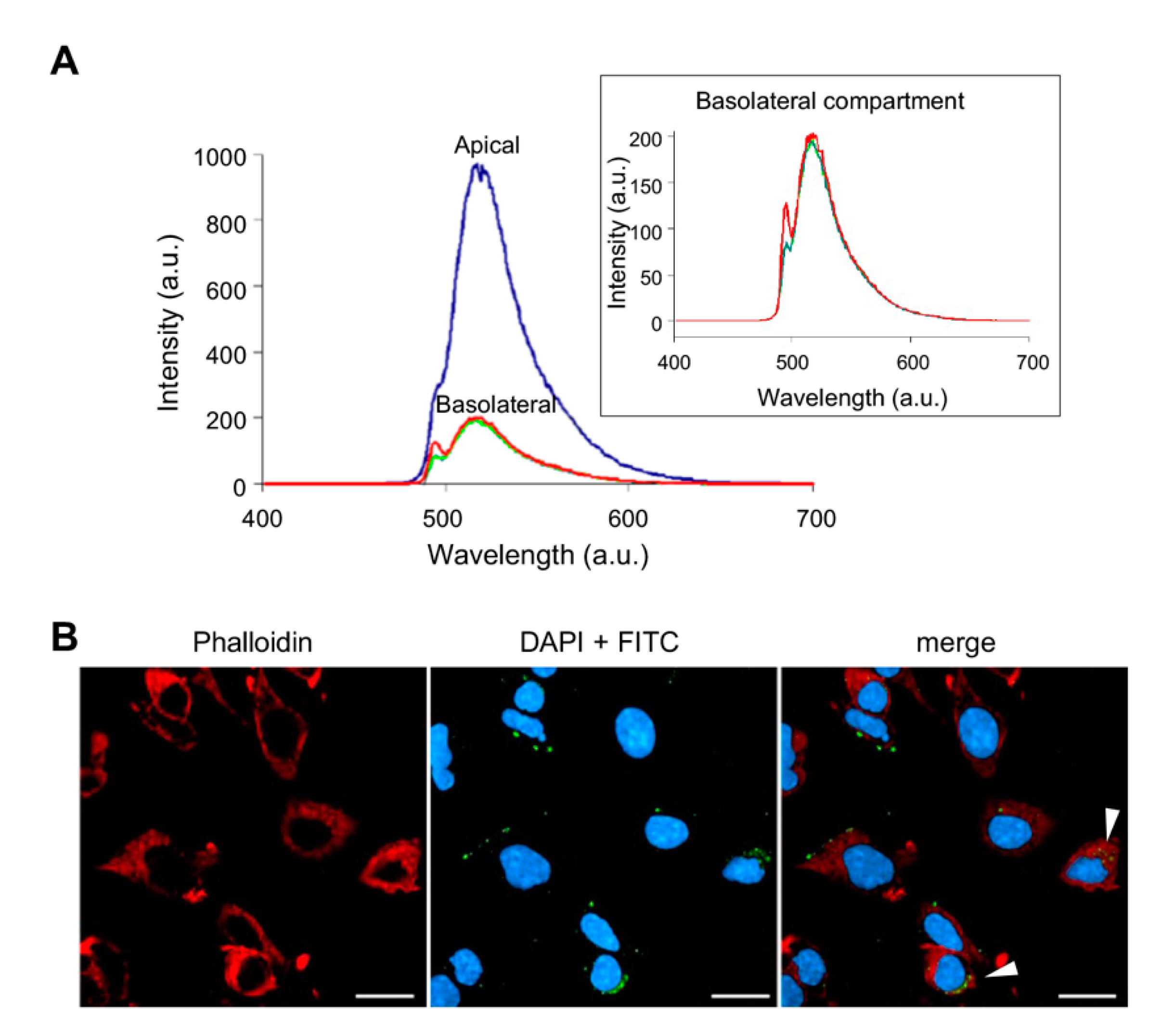
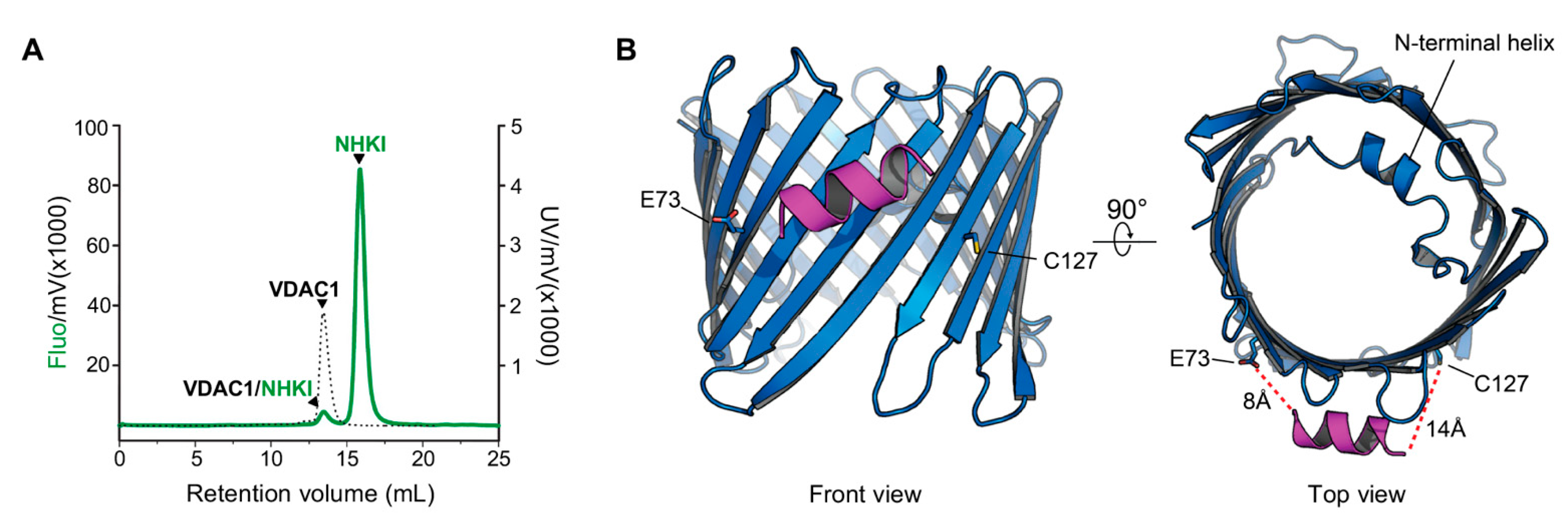
3.1. Assessment of Membrane Permeability to NHK1 Peptide
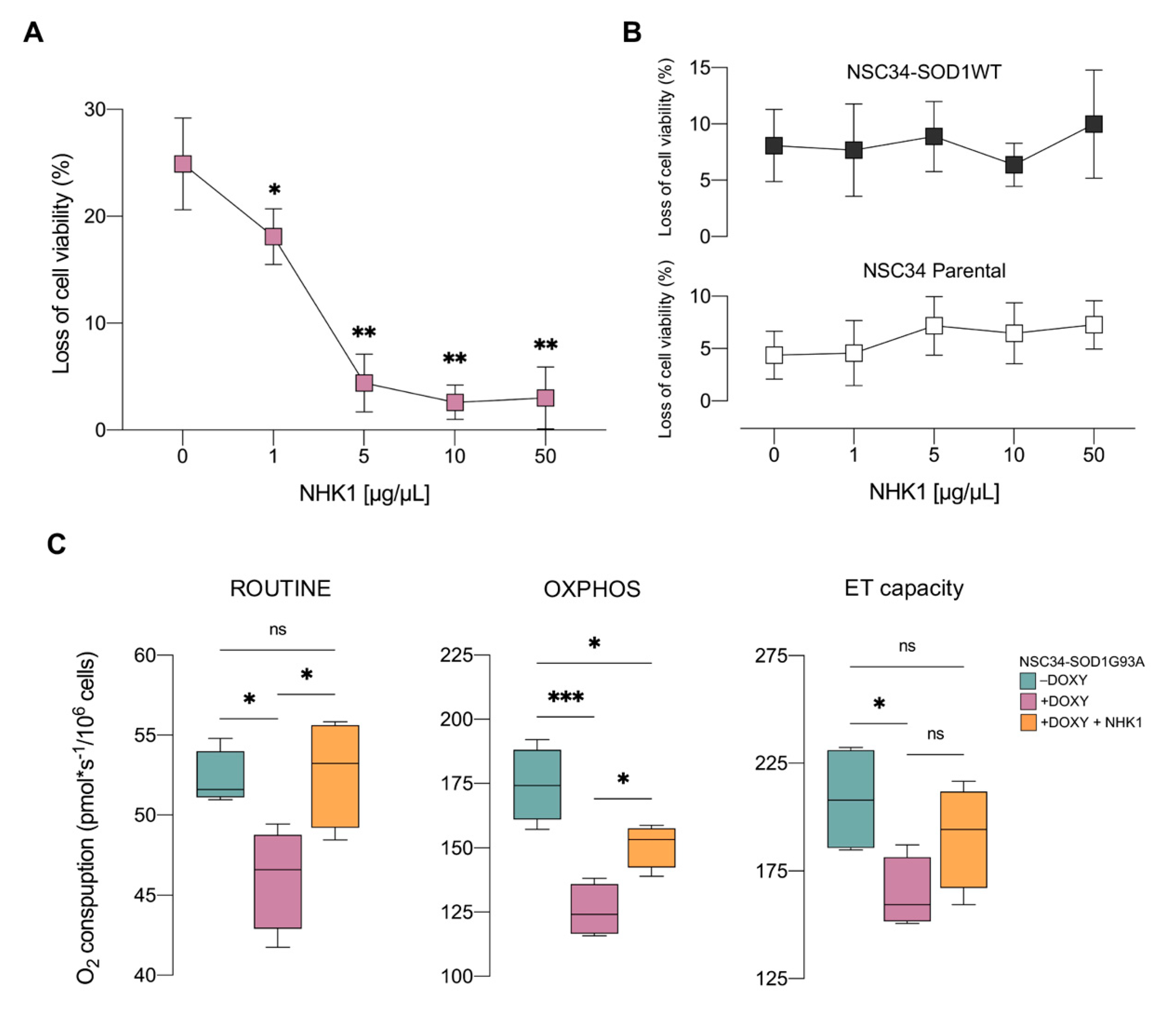
3.2. NHK1 Ameliorates Cell Viability and Oxygen Consumption in NSC34-SOD1G93A Cells
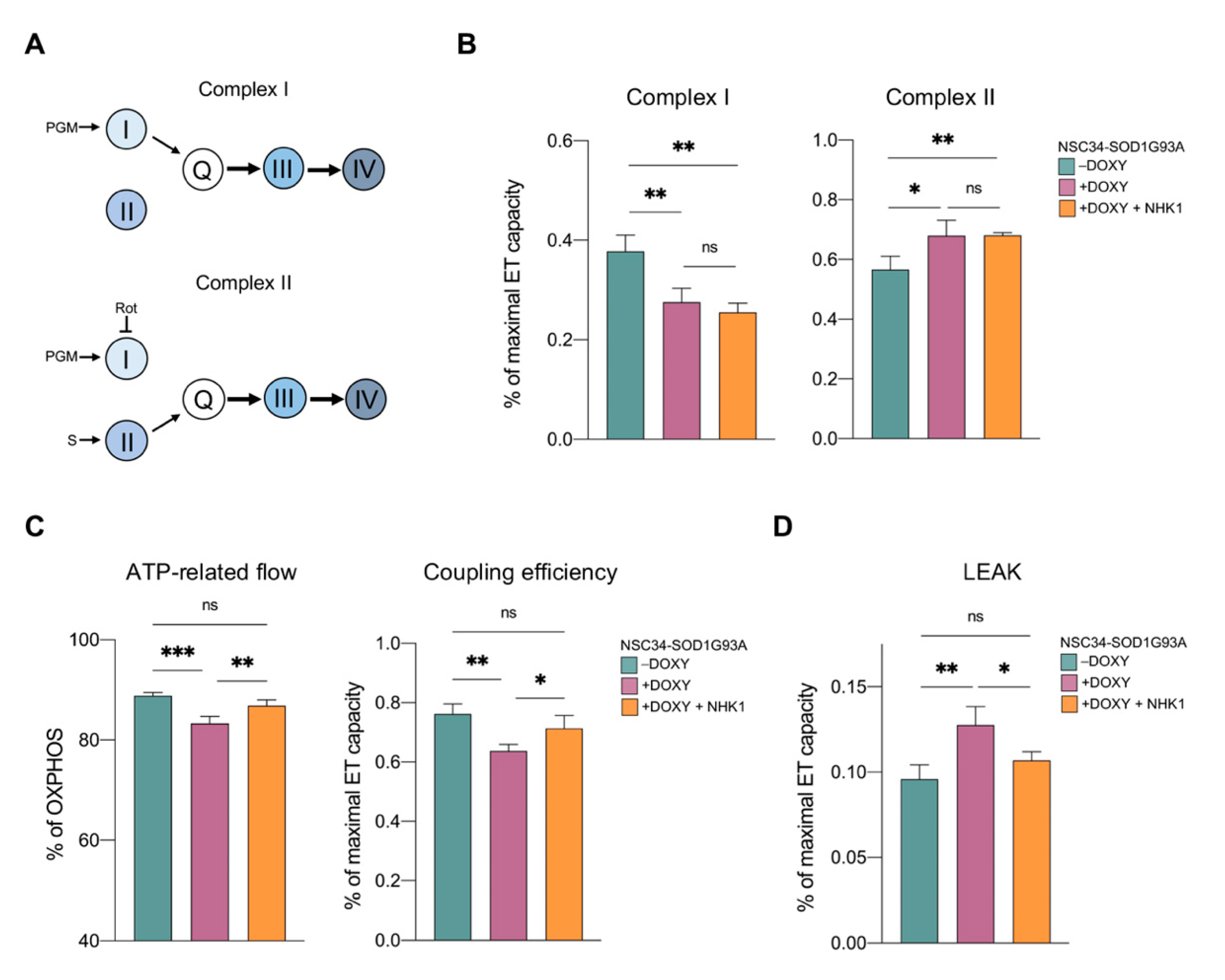
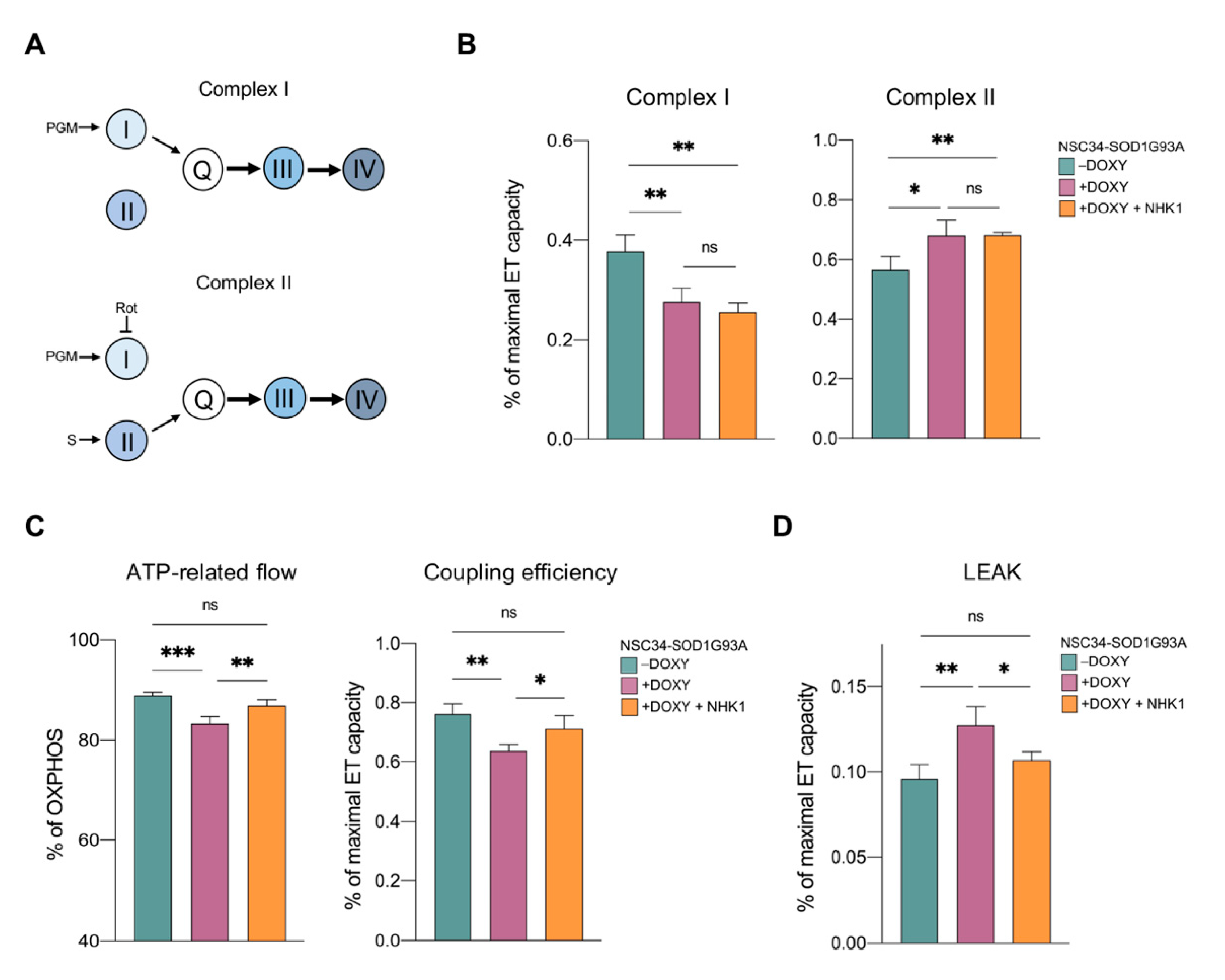
3.3. NHKI Peptide Improves ATP-Linked OXPHOS Flows but Not Complex I Activity
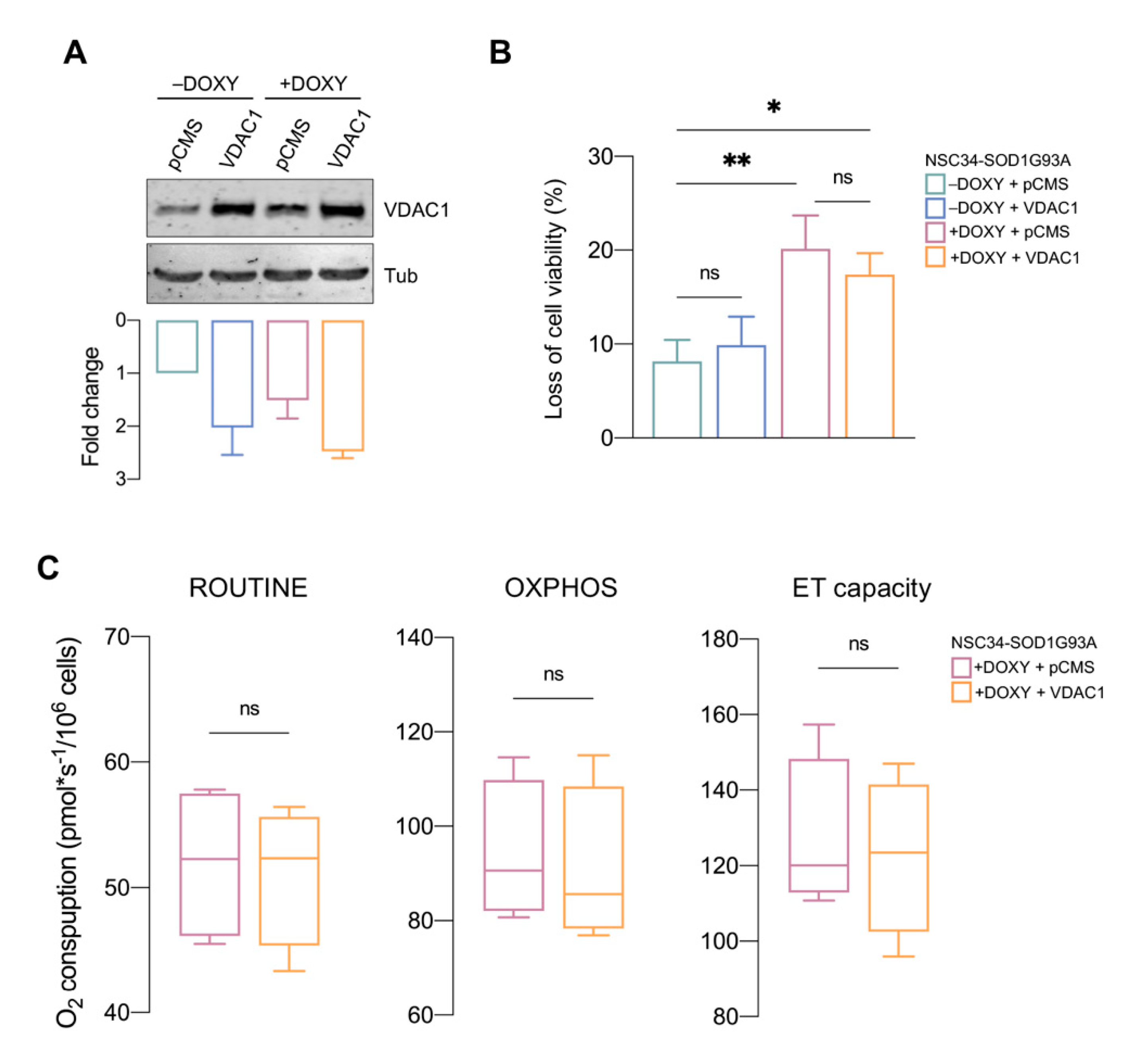
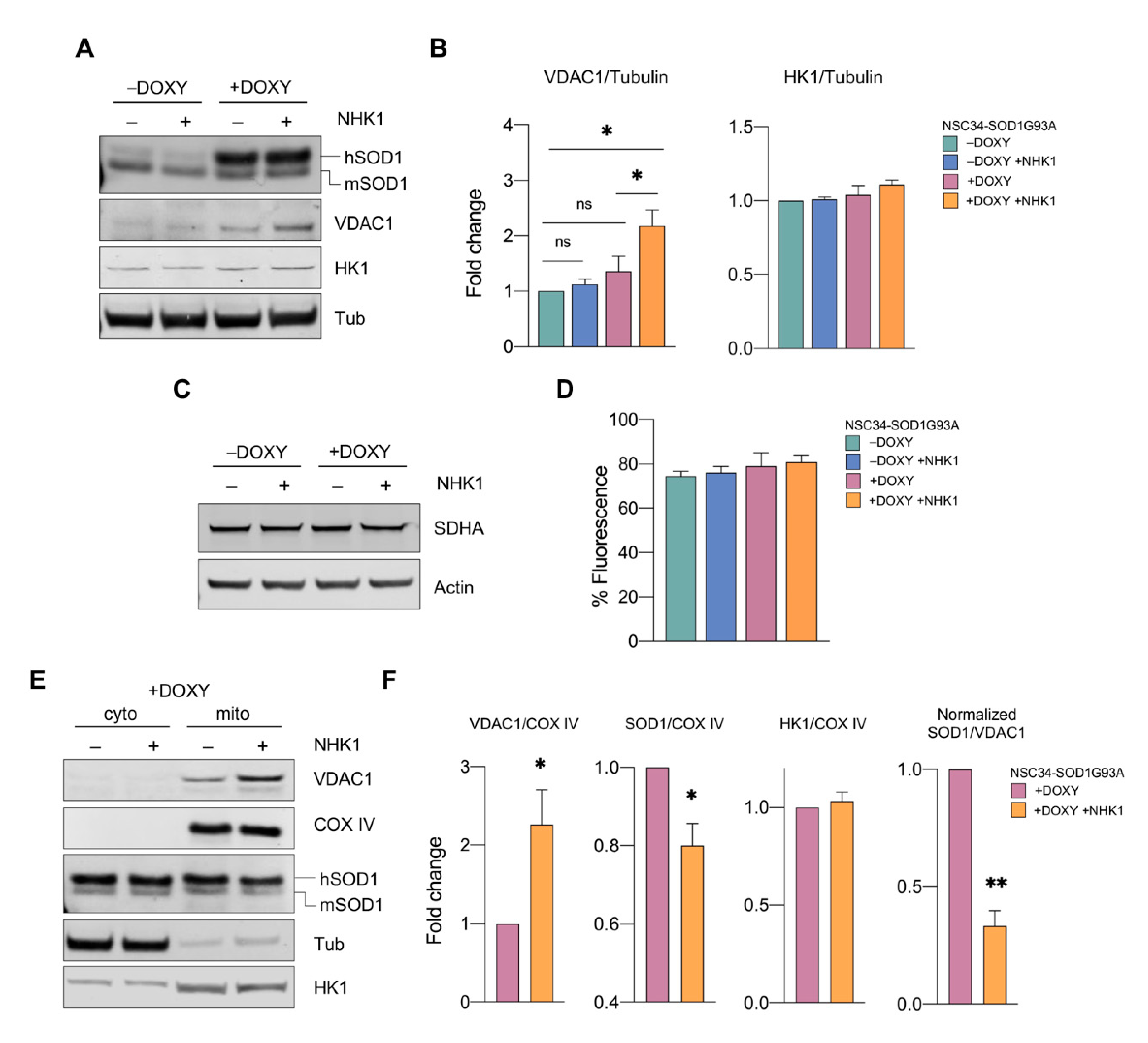
3.4. NHK1 Peptide Increases VDAC1 Levels While Reduces SOD1 G93A Mitochondrial Accumulation
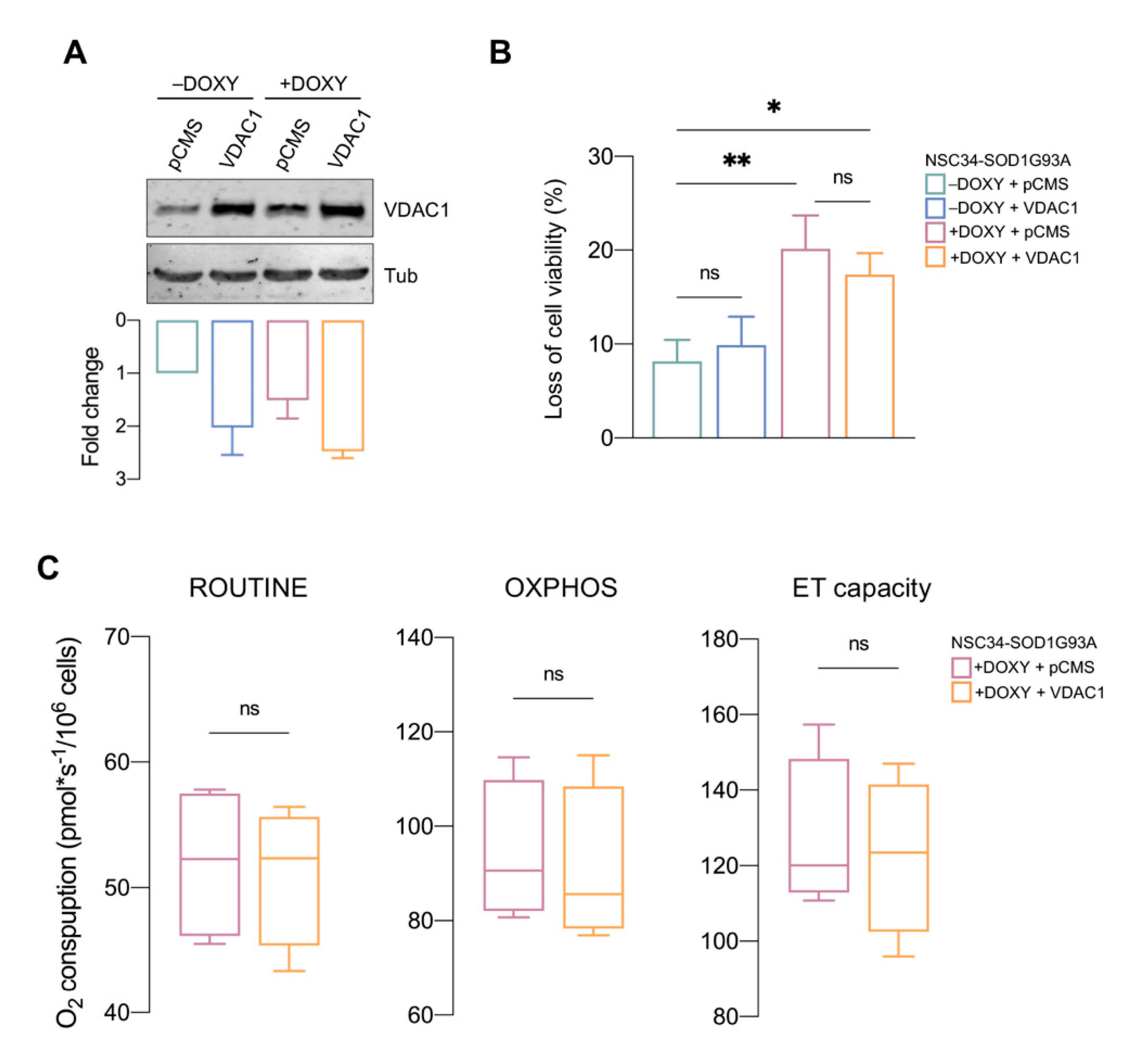
3.5. VDAC1 Overexpression Is Not Sufficient to Counteract SOD1 G93A Toxicity
3.6. NHK1 Peptide Interacts with VDAC1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| DOXY | doxycycline |
| ET | electron transport |
| FCR(s) | flux control ratio(s) |
| FITC | fluoresceine isothiocyanate |
| FSEC | fluorescence-detection size-exclusion chromatography |
| HK | hexokinase |
| HRR | high-resolution respirometry |
| MAMs | mitochondrial-associated membranes |
| MN(s) | motor neuron(s) |
| OMM | outer mitochondrial membrane |
| SOD1 | Cu/Zn superoxide dismutase |
| VDAC | voltage-dependent anion channel |
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The Genetics and Neuropathology of Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Xu, Z.; Hayward, L.J. Aberrantly Increased Hydrophobicity Shared by Mutants of Cu, Zn-Superoxide Dismutase in Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2005, 280, 29771–29779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudencio, M.; Borchelt, D.R. Superoxide Dismutase 1 Encoding Mutations Linked to ALS Adopts a Spectrum of Misfolded States. Mol. Neurodegener. 2011, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zala, D.; Hinckelmann, M.V.; Yu, H.; Da Cunha, M.M.L.; Liot, G.; Cordelières, F.P.; Marco, S.; Saudou, F. Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport. Cell 2013, 152, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Hirano, A.; Donnenfeld, H.; Sasaki, S.; Nakano, I. Fine Structural Observations of Neurofilamentous Changes in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 461–470. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and Respiratory Chain Function in Spinal Cords of ALS Patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussee, E.; de Smet, P.; Bogaert, E.; Elens, I.; van Damme, P.; Willems, P.; Koopman, W.; van den Bosch, L.; Callewaert, G. G37R SOD1 Mutant Alters Mitochondrial Complex I Activity, Ca2+ Uptake and ATP Production. Cell Calcium 2011, 49, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Gurney, M.E. Development of Central Nervous System Pathology in a Murine Transgenic Model of Human Amyotrophic Lateral Sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar] [PubMed]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.A.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial Dysfunction in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrané, J.; Sahawneh, M.A.; Przedborski, S.; Estévez, Á.G.; Manfredi, G. Mitochondrial Dynamics and Bioenergetic Dysfunction Is Associated with Synaptic Alterations in Mutant Sod1 Motor Neurons. J. Neurosci. 2012, 32, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lillo, C.; Jonsson, P.A.; vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of Familial ALS-Linked SOD1 Mutants from Selective Recruitment to Spinal Mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-Superoxide Dismutases Associate with Mitochondria and Shift Their Redox Potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective Association of Misfolded ALS-Linked Mutant SOD1 with the Cytoplasmic Face of Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israelson, A.; Arbel, N.; da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- De Pinto, V. Renaissance of VDAC: New Insights on a Protein Family at the Interface between Mitochondria and Cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Bay, D.C.; Hausner, G.; Court, D.A. The Evolutionary History of Mitochondrial Porins. BMC Evol. Biol. 2007, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiani, C.; Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Bodrenko, I.; Messina, A.; Kmita, H.; Ceccarelli, M.; De Pinto, V. YVDAC2, the Second Mitochondrial Porin Isoform of Saccharomyces Cerevisiae. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 270–279. [Google Scholar] [CrossRef]
- Leggio, L.; Guarino, F.; Magrì, A.; Accardi-Gheit, R.; Reina, S.; Specchia, V.; Damiano, F.; Tomasello, M.F.; Tommasino, M.; Messina, A. Mechanism of Translation Control of the Alternative Drosophila Melanogaster Voltage Dependent Anion-Selective Channel 1 MRNAs. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef] [Green Version]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The Crystal Structure of Mouse VDAC1 at 2.3 Å Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasello, M.F.; Guarino, F.; Reina, S.; Messina, A.; De Pinto, V. The Voltage-Dependent Anion Selective Channel 1 (VDAC1) Topography in the Mitochondrial Outer Membrane as Detected in Intact Cell. PLoS ONE 2013, 8, e81522. [Google Scholar] [CrossRef] [PubMed]
- Benz, R. Permeation of Hydrophilic Solutes through Mitochondrial Outer Membranes: Review on Mitochondrial Porins. BBA Rev. Biomembr. 1994, 1197, 167–196. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a Multi-Functional Mitochondrial Protein Regulating Cell Life and Death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The Voltage-Dependent Anion Channel-1 Modulates Apoptotic Cell Death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I Protection against Apoptotic Cell Death Is Mediated via Interaction with the Voltage-Dependent Anion Channel-1: Mapping the Site of Binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Shimizu, S. VDAC Regulation by the Bcl-2 Family of Proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 Family Proteins Regulate the Release of Apoptogenic Cytochrome c by the Mitochondrial Channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magrì, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrì, A.; Belfiore, R.; Reina, S.; Tomasello, M.F.; Di Rosa, M.C.; Guarino, F.; Leggio, L.; De Pinto, V.; Messina, A. Hexokinase i N-Terminal Based Peptide Prevents the VDAC1-SOD1 G93A Interaction and Re-Establishes ALS Cell Viability. Sci. Rep. 2016, 6, 34802. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In Self-Defence: Hexokinase Promotes Voltage-Dependent Anion Channel Closure and Prevents Mitochondria-Mediated Apoptotic Cell Death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. Ascs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in Vitro Model of Als. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formicola, B.; Dal Magro, R.; Montefusco-Pereira, C.V.; Lehr, C.M.; Koch, M.; Russo, L.; Grasso, G.; Deriu, M.A.; Danani, A.; Bourdoulous, S.; et al. The Synergistic Effect of Chlorotoxin-MApoE in Boosting Drug-Loaded Liposomes across the BBB. J. Nanobiotechnol. 2019, 17, 115. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The HCMEC/D3 Cell Line as a Model of the Human Blood Brain Barrier. Fluids Barriers CNS 2013, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain Barrier-specific Properties of a Human Adult Brain Endothelial Cell Line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Dehouck, B.; Descamps, L.; Fenart, L.; Buée-Scherrer, V.; Duhem, C.; Lundquist, S.; Rentfel, M.; Torpier, G.; Dehouck, M.P. In Vitro Model for Evaluating Drug Transport across the Blood–brain Barrier. Adv. Drug Deliv. Rev. 1999, 36, 165–178. [Google Scholar] [CrossRef]
- Risiglione, P.; Leggio, L.; Cubisino, S.A.M.; Reina, S.; Paternò, G.; Marchetti, B.; Magrì, A.; Iraci, N.; Messina, A. High-Resolution Respirometry Reveals Mpp+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7809. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef]
- Evinova, A.; Cizmarova, B.; Hatokova, Z.; Racay, P. High-Resolution Respirometry in Assessment of Mitochondrial Function in Neuroblastoma SH-SY5Y Intact Cells. J. Membr. Biol. 2020, 253, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger-MitoEagle Task, E. Mitochondrial Physiology Extended Resource of Mitochondrial Respiratory States and Rates. Bioenerg. Commun. 2020, 2020, 1. [Google Scholar] [CrossRef]
- Schindler, A.; Foley, E. Hexokinase 1 Blocks Apoptotic Signals at the Mitochondria. Cell. Signal. 2013, 25, 2685–2692. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Conti Nibali, S.; Messina, A.; Kmita, H.; De Pinto, V. Recombinant Yeast VDAC2: A Comparison of Electrophysiological Features with the Native Form. FEBS Open Bio 2019, 9, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Reina, S.; Checchetto, V.; Saletti, R.; Gupta, A.; Chaturvedi, D.; Guardiani, C.; Guarino, F.; Scorciapino, M.A.; Magrì, A.; Foti, S.; et al. VDAC3 as a Sensor of Oxidative State of the Intermembrane Space of Mitochondria: The Putative Role of Cysteine Residue Modifications. Oncotarget 2016, 7, 2249–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawate, T.; Gouaux, E. Fluorescence-Detection Size-Exclusion Chromatography for Precrystallization Screening of Integral Membrane Proteins. Structure 2006, 14, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- Chang, K.Y.; Yang, J.R. Analysis and Prediction of Highly Effective Antiviral Peptides Based on Random Forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef] [Green Version]
- Maji, D.; Lu, J.; Sarder, P.; Schmieder, A.H.; Cui, G.; Yang, X.; Pan, D.; Achilefu, S.; Lanza, G.M. Cellular Trafficking of Sn-2 Phosphatidylcholine Prodrugs Studied with Fluorescence Lifetime Imaging and Super-Resolution Microscopy. Precis. Nanomed. 2018, 1, 128. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular Localization of VDAC in Mitochondria and ER in the Cerebellum. Biochim. Biophys. Acta Bioenerg. 2004, 1657, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, D.; Ramírez-Núñez, O.; Granado-Serrano, A.B.; Torres, P.; Ayala, V.; Moiseeva, V.; Povedano, M.; Ferrer, I.; Pamplona, R.; Portero-Otin, M.; et al. Early and Gender-Specific Differences in Spinal Cord Mitochondrial Function and Oxidative Stress Markers in a Mouse Model of ALS. Acta Neuropathol. Commun. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, E.; Perl, A. Measurement of Mitochondrial Mass by Flow Cytometry during Oxidative Stress. React. Oxyg. Species 2017, 4, 275. [Google Scholar] [CrossRef] [Green Version]
- Pittalà, M.G.G.; Reina, S.; Cubisino, S.A.M.; Cucina, A.; Formicola, B.; Cunsolo, V.; Foti, S.; Saletti, R.; Messina, A. Post-Translational Modification Analysis of Vdac1 in Als-Sod1 Model Cells Reveals Specific Asparagine and Glutamine Deamidation. Antioxidants 2020, 9, 1218. [Google Scholar] [CrossRef]
- Li, Q.; vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-Linked Mutant Superoxide Dismutase 1 (SOD1) Alters Mitochondrial Protein Composition and Decreases Protein Import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef] [Green Version]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic Lateral Sclerosis-Associated SOD1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC Isoforms in Mammals. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by SiRNA Inhibits Cancer Cell Proliferation and Tumor Growth in Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Head, S.A.; Shi, W.; Zhao, L.; Gorshkov, K.; Pasunooti, K.; Chen, Y.; Deng, Z.; Li, R.J.; Shim, J.S.; Tan, W.; et al. Antifungal Drug Itraconazole Targets VDAC1 to Modulate the AMPK/MTOR Signaling Axis in Endothelial Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7276–E7285. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Di Rosa, M.C.; Orlandi, I.; Guarino, F.; Reina, S.; Guarnaccia, M.; Morello, G.; Spampinato, A.; Cavallaro, S.; Messina, A.; et al. Deletion of Voltage-Dependent Anion Channel 1 Knocks Mitochondria down Triggering Metabolic Rewiring in Yeast. Cell. Mol. Life Sci. 2020, 77, 3195–3213. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 Selectively Transfers Apoptotic Ca 2 Signals to Mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. MAM (Mitochondria-Associated Membranes) in Mammalian Cells: Lipids and Beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated Membrane Collapse Is a Common Pathomechanism in SIGMAR 1—And SOD 1—Linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Le Verche, V.; Przedborski, S. Is Amyotrophic Lateral Sclerosis a Mitochondrial Channelopathy? Neuron 2010, 67, 523–524. [Google Scholar] [CrossRef] [Green Version]
- Serviddio, G.; Sastre, J. Measurement of Mitochondrial Membrane Potential and Proton Leak. Methods Mol. Biol. 2010, 594, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.K.; Godbole, A.; Mathew, M.K. Regulation of VDAC Trafficking Modulates Cell Death. Cell Death Discov. 2016, 2, 16085. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-Regulation of Hexokinase II Contributes to Rituximabchemotherapy Resistance and Is a Clinically Relevant Target for Therapeutic Development. Oncotarget 2018, 9, 4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide Dismutase 1 Acts as a Nuclear Transcription Factor to Regulate Oxidative Stress Resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrì, A.; Di Rosa, M.C.; Tomasello, M.F.; Guarino, F.; Reina, S.; Messina, A.; De Pinto, V. Overexpression of Human SOD1 in VDAC1-Less Yeast Restores Mitochondrial Functionality Modulating Beta-Barrel Outer Membrane Protein Genes. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Pathak, G.A.; Zhou, Z.; Silzer, T.K.; Barber, R.C.; Phillips, N.R. Two-Stage Bayesian GWAS of 9576 Individuals Identifies SNP Regions That Are Targeted by MiRNAs Inversely Expressed in Alzheimer’s and Cancer. Alzheimer’s Dement. 2020, 16, 162–177. [Google Scholar] [CrossRef]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, X.E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting Vdac1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.Y.; Kim, Y.R.; Choi, K.W.; Lee, M.; Lee, S.; Im, W.; Shin, J.Y.; Kim, J.Y.; Hong, Y.H.; Kim, M.; et al. Downregulated MiR-18b-5p Triggers Apoptosis by Inhibition of Calcium Signaling and Neuronal Cell Differentiation in Transgenic SOD1 (G93A) Mice and SOD1 (G17S and G86S) ALS Patients. Transl. Neurodegener. 2020, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Guarino, F.; Zinghirino, F.; Mela, L.; Pappalardo, X.G.; Ichas, F.; De Pinto, V.; Messina, A. NRF-1 and HIF-1α Contribute to Modulation of Human VDAC1 Gene Promoter during Starvation and Hypoxia in HeLa Cells. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148289. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I Activity Signals Antioxidant Response through ERK5. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Guarino, F.; De Pinto, V. Is the Secret of Vdac Isoforms in Their Gene Regulation? Characterization of Human Vdac Genes Expression Profile, Promoter Activity, and Transcriptional Regulators. Int. J. Mol. Sci. 2020, 21, 7388. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage-Dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of VDAC1 with Amyloid Beta and Phosphorylated Tau Causes Mitochondrial Dysfunction in Alzheimer’s Disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Magri, A.; Messina, A. Interactions of VDAC with Proteins Involved in Neurodegenerative Aggregation: An Opportunity for Advancement on Therapeutic Molecules. Curr. Med. Chem. 2017, 24, 4470–4487. [Google Scholar] [CrossRef]
- Risiglione, P.; Zinghirino, F.; Di Rosa, M.C.; Magrì, A.; Messina, A. Alpha-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease: The Emerging Role of Vdac. Biomolecules 2021, 11, 718. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magrì, A.; Risiglione, P.; Caccamo, A.; Formicola, B.; Tomasello, M.F.; Arrigoni, C.; Zimbone, S.; Guarino, F.; Re, F.; Messina, A. Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration. Biomedicines 2021, 9, 948. https://doi.org/10.3390/biomedicines9080948
Magrì A, Risiglione P, Caccamo A, Formicola B, Tomasello MF, Arrigoni C, Zimbone S, Guarino F, Re F, Messina A. Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration. Biomedicines. 2021; 9(8):948. https://doi.org/10.3390/biomedicines9080948
Chicago/Turabian StyleMagrì, Andrea, Pierpaolo Risiglione, Antonella Caccamo, Beatrice Formicola, Marianna Flora Tomasello, Cristina Arrigoni, Stefania Zimbone, Francesca Guarino, Francesca Re, and Angela Messina. 2021. "Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration" Biomedicines 9, no. 8: 948. https://doi.org/10.3390/biomedicines9080948
APA StyleMagrì, A., Risiglione, P., Caccamo, A., Formicola, B., Tomasello, M. F., Arrigoni, C., Zimbone, S., Guarino, F., Re, F., & Messina, A. (2021). Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration. Biomedicines, 9(8), 948. https://doi.org/10.3390/biomedicines9080948