
Heterogeneous Types of miRNA-Disease Associations Stratified by Multi-Layer Network Embedding and Prediction


Abstract
:1. Introduction
2. Materials and Methods
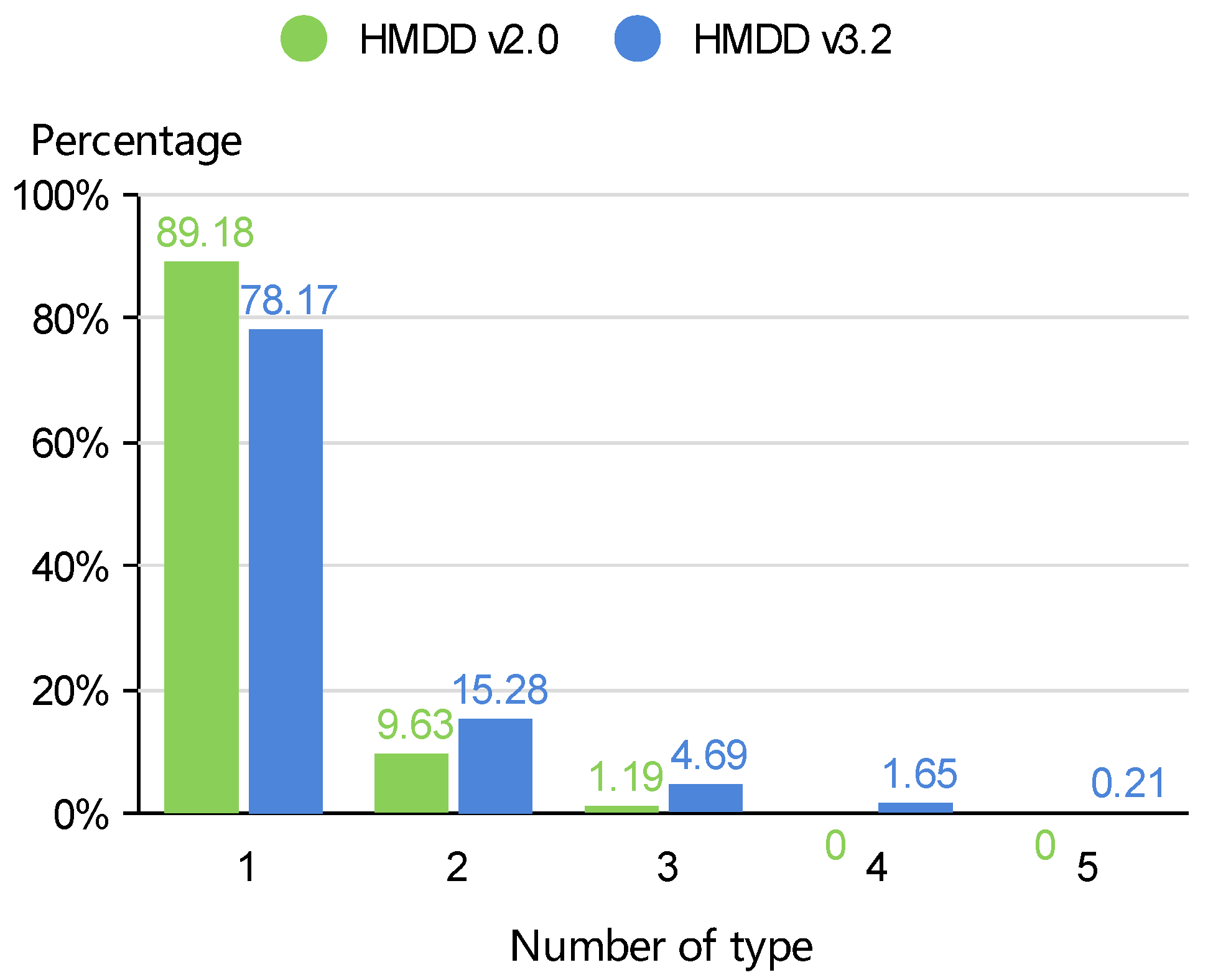
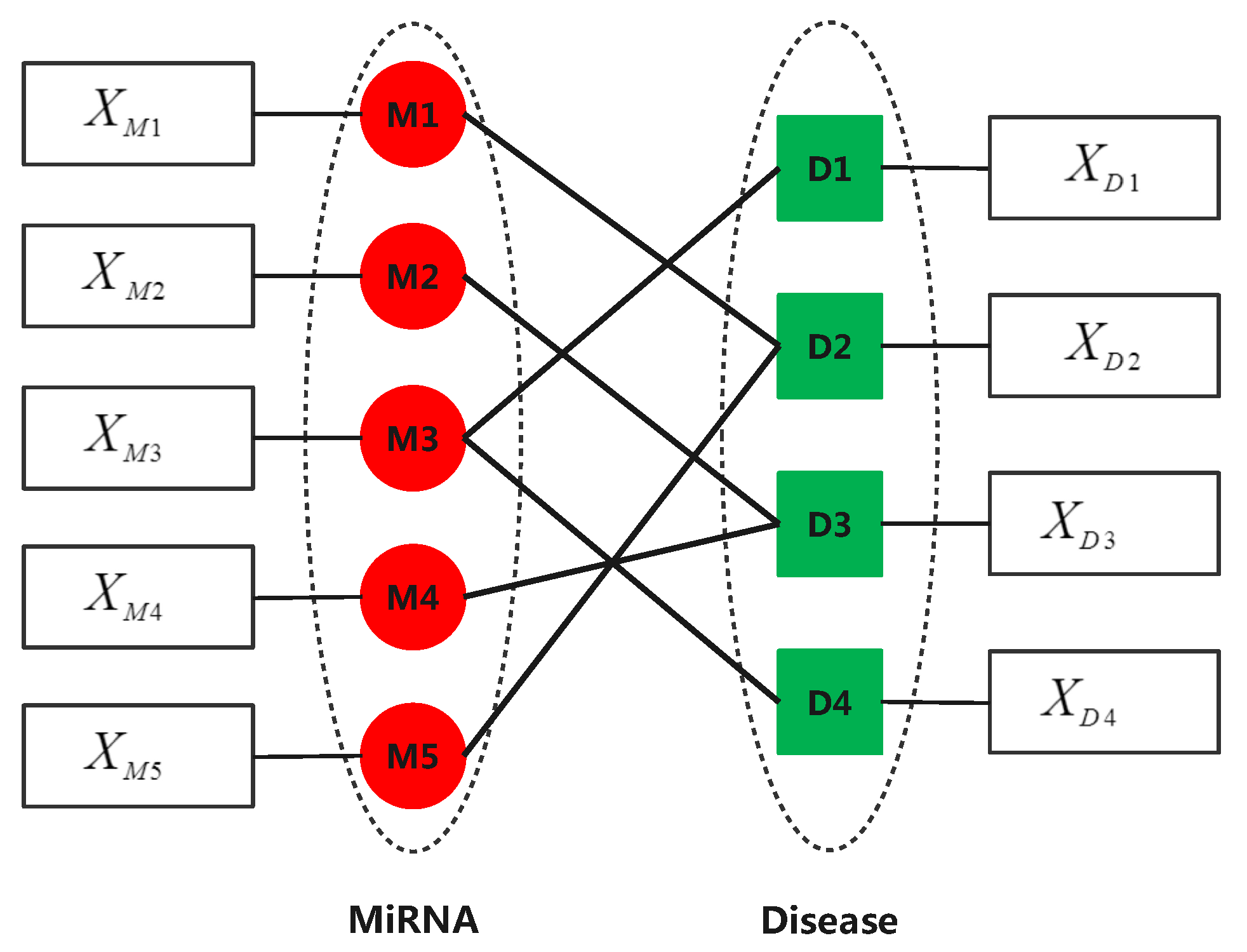
2.1. Datasets
2.2. Similarity Calculation for Diseases and miRNAs
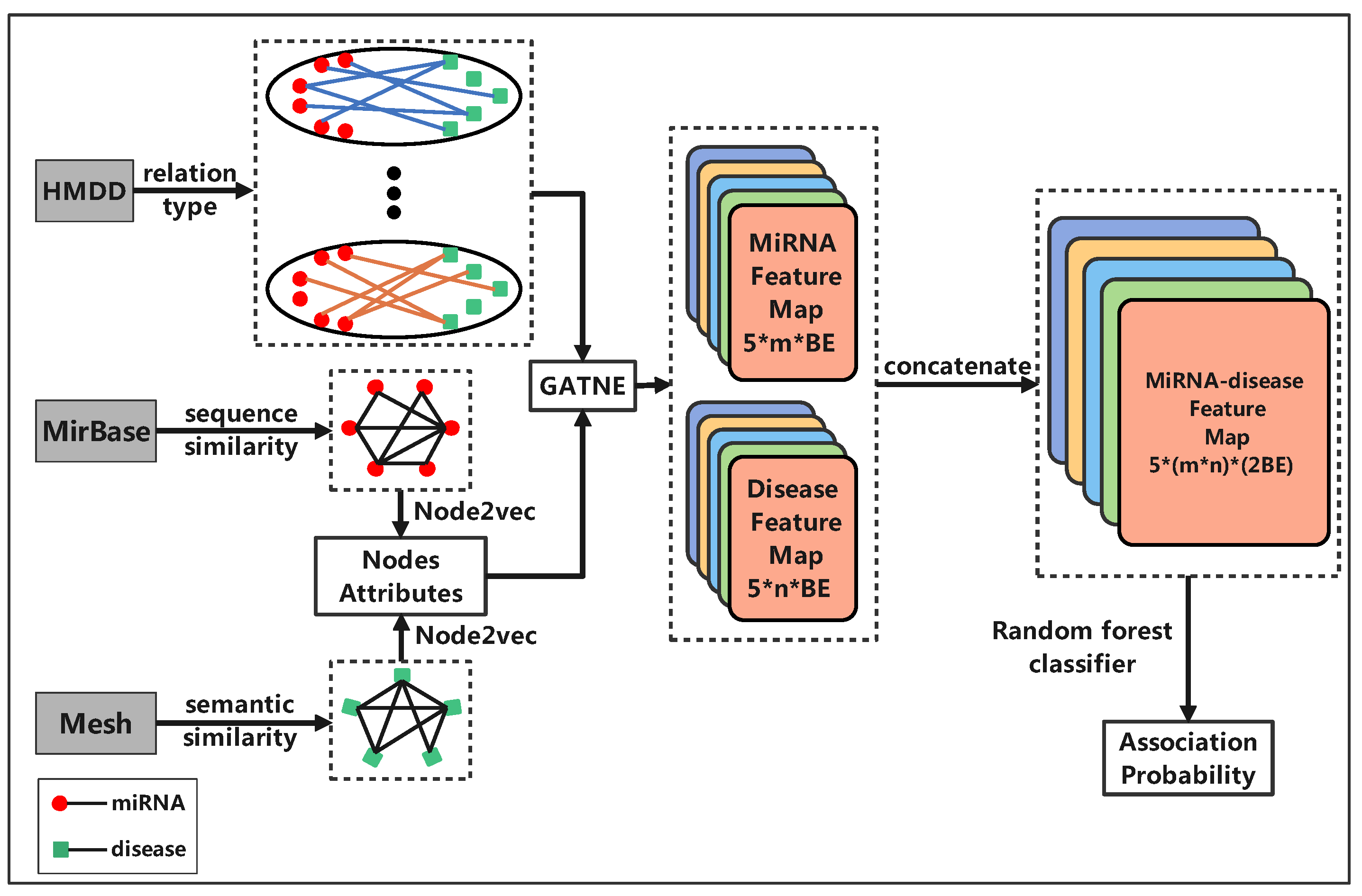
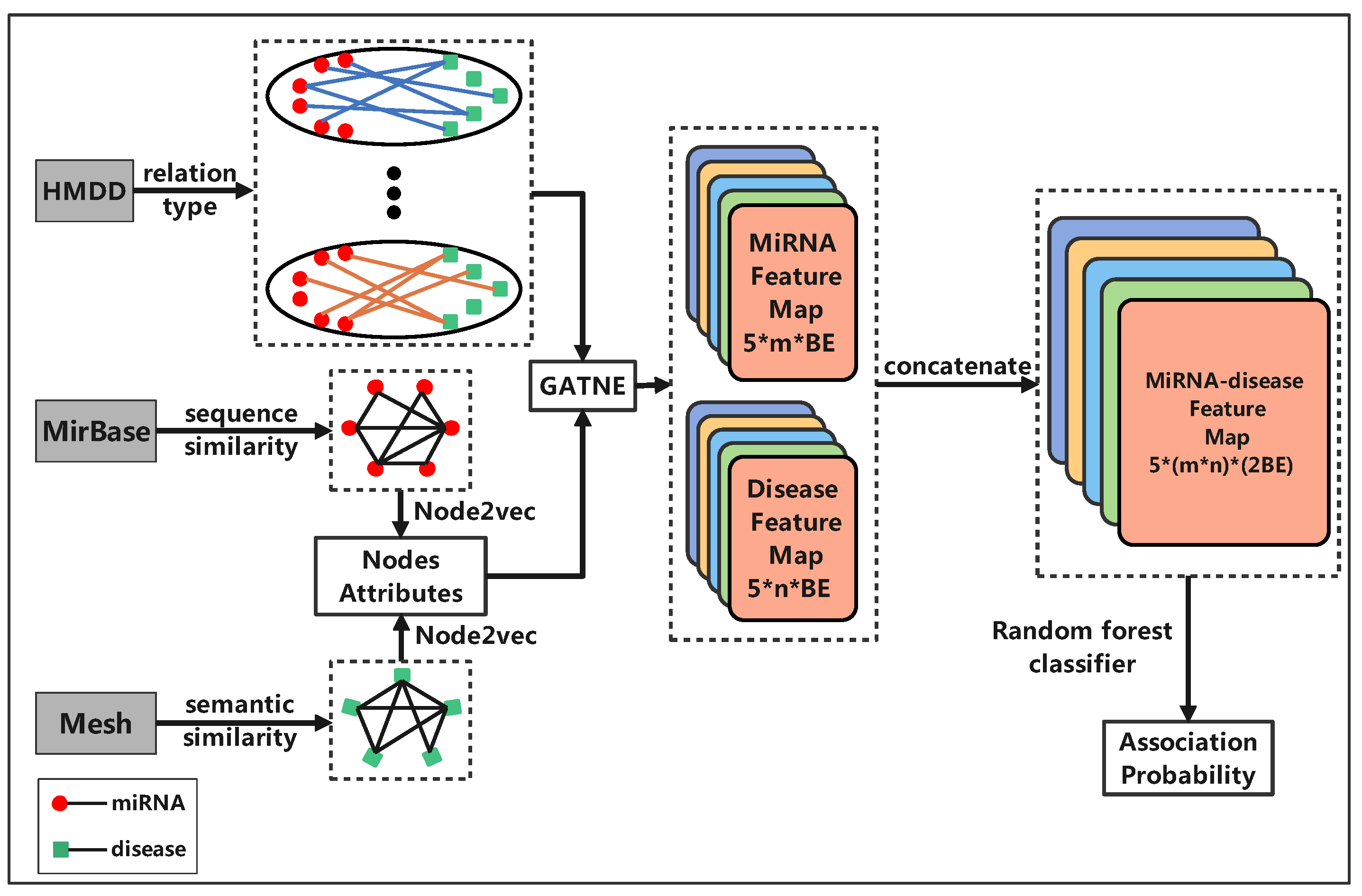
2.3. Graph Embedding
2.3.1. Node2vec
2.3.2. GATNE
2.4. mDLinker
3. Experiments and Results
3.1. Experimental Setting
3.2. Performance by Different Classifiers
3.3. Performance Comparison with the State of the Art
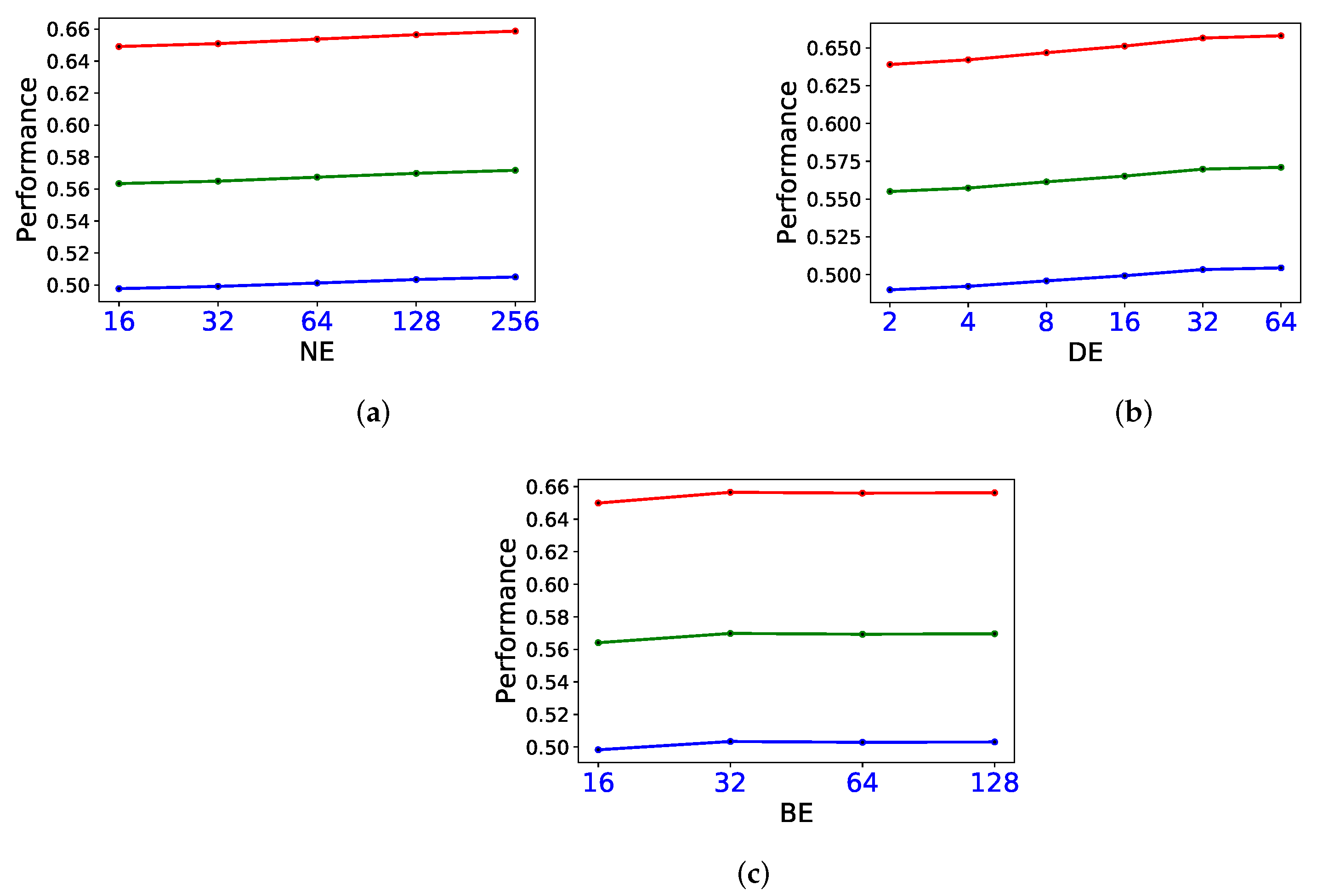
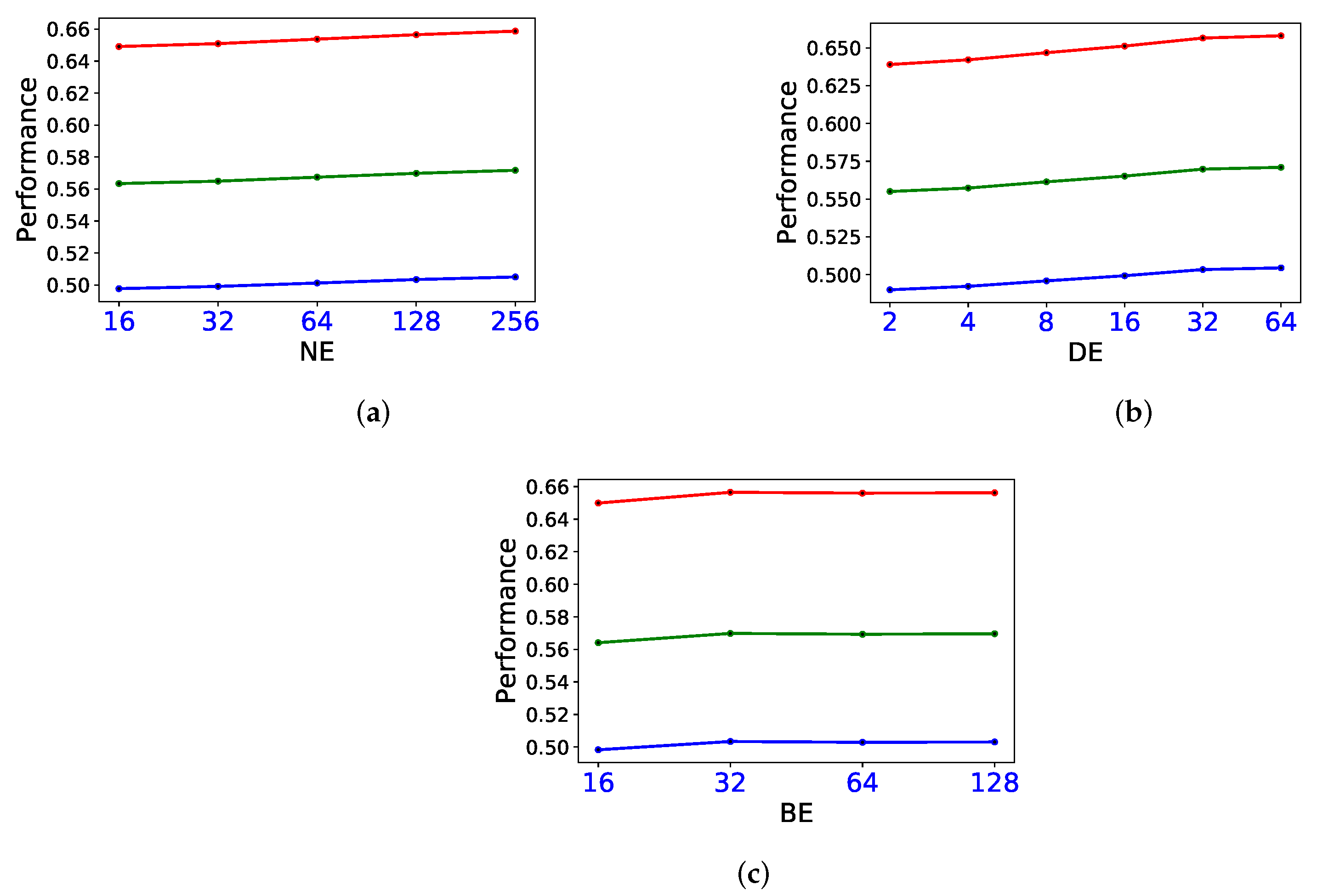
3.4. Sensitivity Study on Parameters
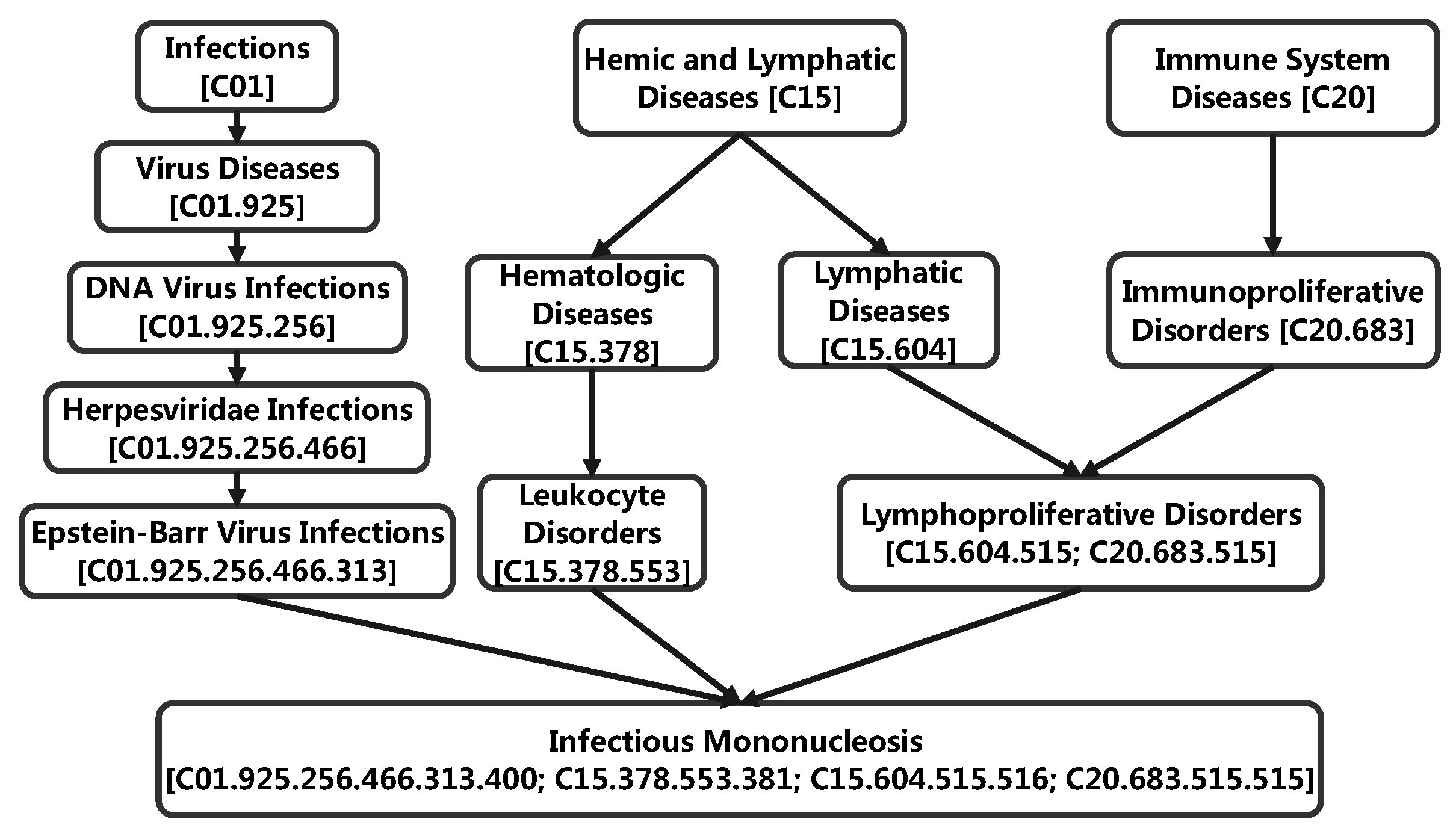
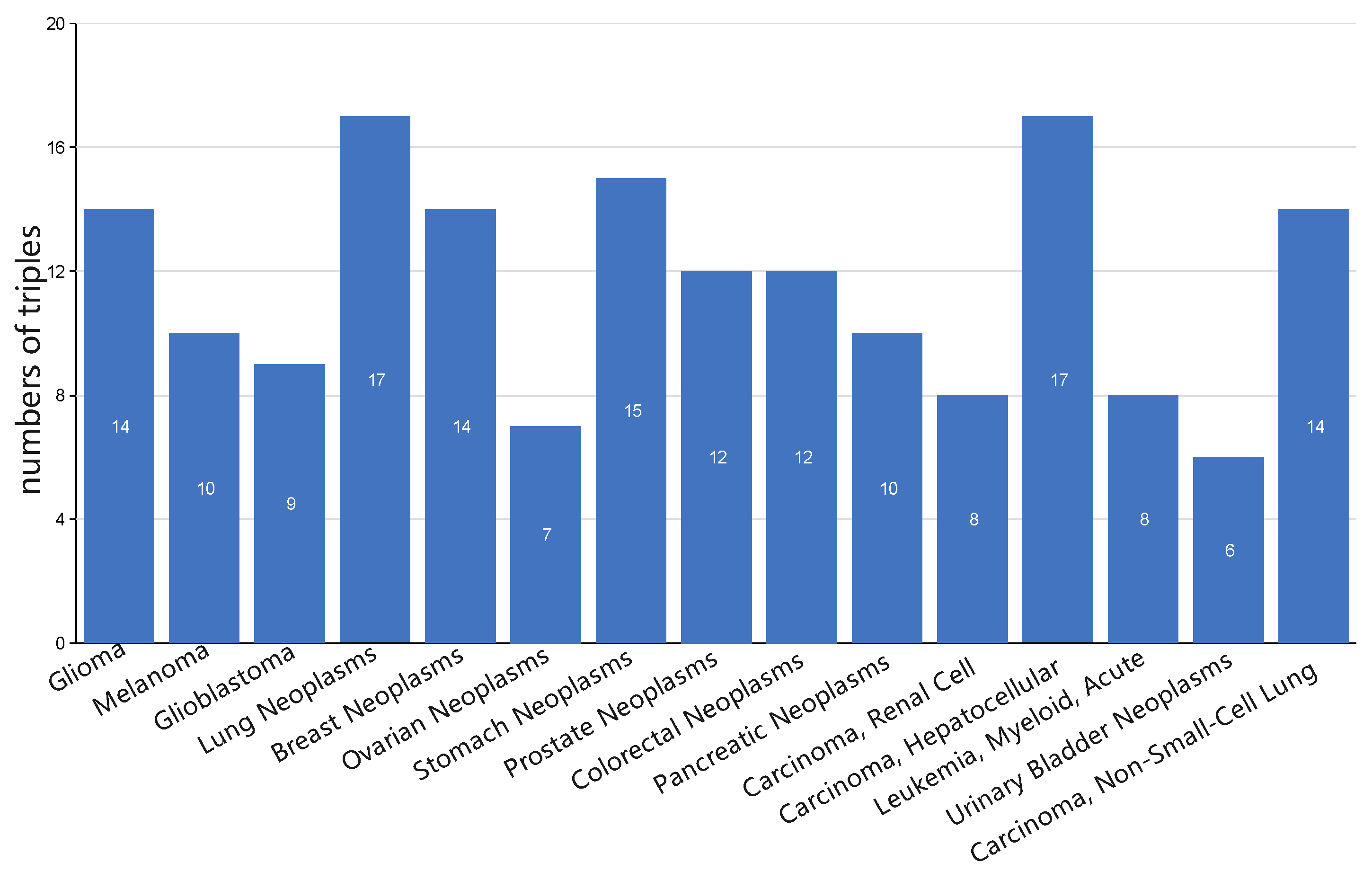
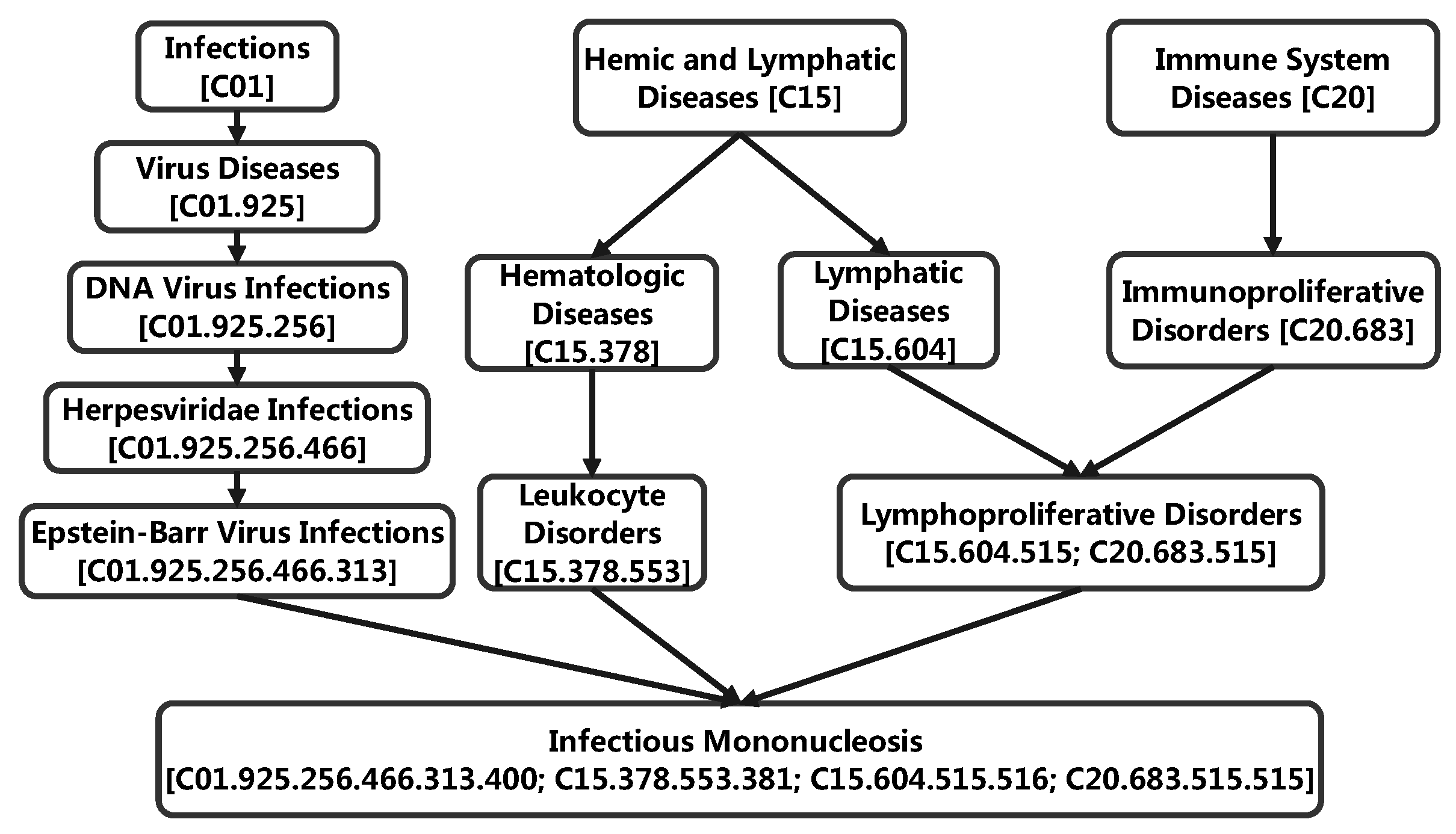
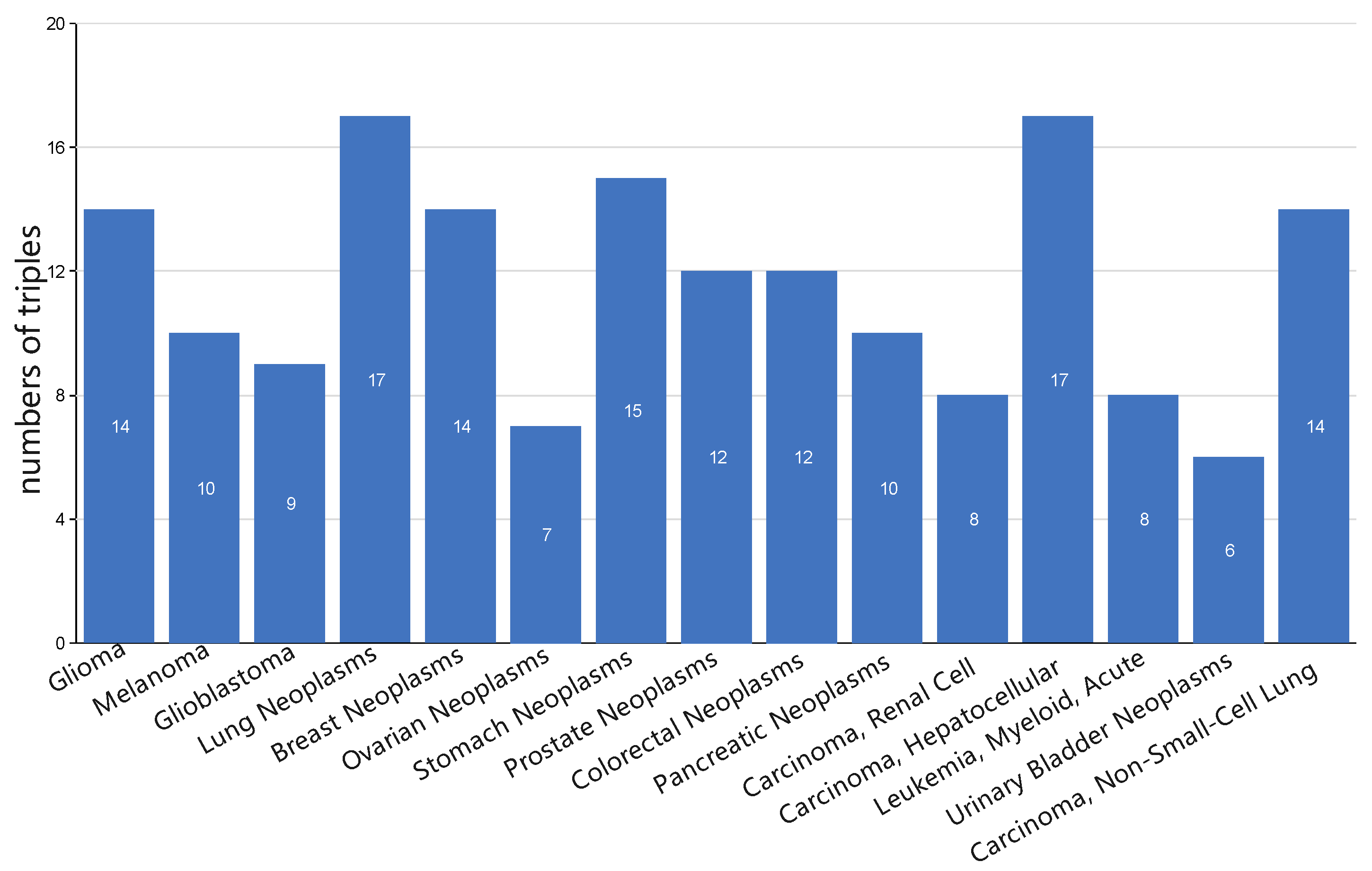
3.5. Case Study: miRNA-Disease Association Types Predicted beyond the HMDD Databases
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HMDD | miRNA | Disease | MD-P a | MD-T b | ||||
|---|---|---|---|---|---|---|---|---|
| Type-1 | Type-2 | Type-3 | Type-4 | Type-5 | ||||
| v2.0 | 321 | 168 | 1506 | 355 | 215 | 441 | 676 | 0 |
| v3.2 | 695 | 445 | 12,495 | 1578 | 519 | 3300 | 5822 | 5079 |
References
- Lynam-Lennon, N.; Maher, S.G.; Reynolds, J.V. The roles of microRNA in cancer and apoptosis. Biol. Rev. Camb. Philos. Soc. 2009, 84, 55–71. [Google Scholar] [CrossRef]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Feng, Y.; Coukos, G.; Zhang, L. Therapeutic microRNA strategies in human cancer. AAPS J. 2009, 11, 747–757. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2019, 20, 515–539. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, C.M.; Qi, L.; He, T.Z.; Shi-Guo, L.; Hao, C.J.; Ma, X. Two common SNPs in pri-miR-125a alter the mature miRNA expression and associate with recurrent pregnancy loss in a Han-Chinese population. RNA Biol. 2011, 8, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lu, S.; Wang, Y.; Guo, S.; Zhao, T.; Wang, X.; Song, B. Influence of microRNA 34a on proliferation, invasion and metastasis of HCT116 cells. Mol. Med. Rep. 2017, 15, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Schein, J.E. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Tewari, M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, Q.; Zhang, R.; Dai, X.; Chen, W.; Xing, D. Circulating microRNAs: Biomarkers of disease. Clin. Chim. Acta 2021, 516, 46–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Zhang, C.Y. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Rao, Y.M.; Shi, H.R.; Ji, M.; Chen, C.H. MiR-106a targets Mcl-1 to suppress cisplatin resistance of ovarian cancer A2780 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 567–572. [Google Scholar] [CrossRef]
- Zu, L.; Xue, Y.; Wang, J.; Fu, Y.; Wang, X.; Xiao, G.; Wang, J. The feedback loop between miR-124 and TGF-β pathway plays a significant role in non-small cell lung cancer metastasis. Carcinogenesis 2016, 37, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Zhou, L.; Wang, H.; Yang, Y.; Sun, Y.; Wang, Z.; Li, H. Differential expression profiles of microRNAs in highly and weakly invasive/metastatic pancreatic cancer cells. Oncol. Lett. 2018, 16, 6026–6038. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Cui, Q. The relationship of human tissue microRNAs with those from body fluids. Sci. Rep. 2020, 10, 5644. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, C.C.; Zhang, X.; Li, Z.; Deng, L.; Zhang, Y.; Dai, Q. RBMMMDA: Predicting multiple types of disease-microRNA associations. Sci. Rep. 2015, 5, 13877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yin, J.; Zhang, X. A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network. Genes 2018, 9, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Yue, X.; Xiong, Z.; Yu, Z.; Liu, S.; Zhang, W. Tensor decomposition with relational constraints for predicting multiple types of microRNA-disease associations. Brief. Bioinform. 2020, 22, bbaa140. [Google Scholar]
- Cen, Y.K.; Zou, X.; Zhang, J.W.; Yang, H.X.; Zhou, J.G.; Tang, J. Representation Learning for Attributed Multiplex Heterogeneous Network. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (KDD 2019), Anchorage, AK, USA, 4–8 August 2019; pp. 1358–1368. [Google Scholar]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; You, Z.H.; Wang, L.; Yan, X.; Li, Z.W. DF-MDA: An effective diffusion-based computational model for predicting miRNA-disease association. Mol. Ther. 2021, 29, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.Y.; You, Z.H.; Wang, Y.; Li, Z.W.; Wong, L. DANE-MDA: Predicting microRNA-disease associations via deep attributed network embedding. iScience 2021, 24, 102455. [Google Scholar] [CrossRef] [PubMed]
- Che, K.; Guo, M.; Wang, C.; Liu, X.; Chen, X. Predicting MiRNA-Disease Association by Latent Feature Extraction with Positive Samples. Genes 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; You, Z.H.; Wang, L.; Zhou, Y.; Li, L.P.; Li, Z.W. DBMDA: A Unified Embedding for Sequence-Based miRNA Similarity Measure with Applications to Predict and Validate miRNA-Disease Associations. Mol. Ther. Nucleic Acids 2020, 19, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Leskovec, J. Node2vec: Scalable Feature Learning for Networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD 2016), San Francisco, CA, USA, 13–17 August 2016; pp. 855–864. [Google Scholar]
- Hamilton, W.; Ying, Z.T.; Leskovec, J. Inductive representation learning on large graphs. In Proceedings of the 31st International Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 1024–1034. [Google Scholar]
- Lin, Z.H.; Feng, M.W.; Santos, C.N.; Yu, M.; Xiang, B.; Zhou, B.; Bengio, Y. A Structured Self-attentive Sentence Embedding. In Proceedings of the International Conference on Learning Representations, Toulon, France, 24–26 April 2017. [Google Scholar]
- Dong, Y.X.; Chawla, N.V.; Swami, A. Metapath2vec: Scalable Representation Learning for Heterogeneous Networks. In Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD 2017), Halifax, NS, Canada, 13–17 August 2017; pp. 135–144. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.S.; Dean, J. Efficient Estimation of Word Representations in Vector Space. In Proceedings of the Workshop at ICLR, Scottsdale, AZ, USA, 2–4 May 2013. [Google Scholar]
- Li, C.; Liu, H.; Hu, Q.; Que, J.; Yao, J. A Novel Computational Model for Predicting microRNA-Disease Associations Based on Heterogeneous Graph Convolutional Networks. Cells 2019, 8, 977. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Li, Y.; Wu, F.X. Enhancing the prediction of disease-gene associations with multimodal deep learning. Bioinformatics 2019, 35, 3735–3742. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, L.; Gao, Y.; Shi, J.; Cui, Q.; Li, J.; Zhou, Y. Benchmark of computational methods for predicting microRNA-disease associations. Genome Biol. 2019, 20, 202. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Dong, Y.; Yang, R.; Wei, L. LINC00619 restricts gastric cancer progression by preventing microRNA-224-5p-mediated inhibition of OPCML. Arch. Biochem. Biophys. 2020, 689, 108390. [Google Scholar] [CrossRef]
- Kojima, K.; Fujita, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J. Biol. Chem. 2010, 285, 19076–19084. [Google Scholar]


| Method | Top-1 Precision | Top-1 Recall | Top-1 F1 |
|---|---|---|---|
| Decision tree | 0.4203 | 0.3224 | 0.3649 |
| Naive Bayes | 0.4748 | 0.3640 | 0.4121 |
| Logistic Regression | 0.4960 | 0.3803 | 0.4305 |
| KNN | 0.6001 | 0.4602 | 0.5209 |
| SVM | 0.5706 | 0.4357 | 0.4952 |
| Random Forest | 0.6509 | 0.4991 | 0.5649 |
| Method | Top-1 Precision | Top-1 Recall | Top-1 F1 |
|---|---|---|---|
| TFAI | 0.5874 | 0.4501 | 0.4832 |
| TDRC | 0.6116 | 0.4686 | 0.5071 |
| mDLinker | 0.6509 | 0.4991 | 0.5649 |
| Method | AUPR | AUC | F1 |
|---|---|---|---|
| TFAI | 0.9261 | 0.912 | 0.8559 |
| TDRC | 0.93 | 0.9222 | 0.865 |
| mDLinker | 0.9799 | 0.9737 | 0.9291 |
| MiRNA | Disease | Type | PMID | Experimental Methods | Description |
|---|---|---|---|---|---|
| hsa-mir-224 | Gastric Neoplasms | Target | 32359894 | RT-qPCR; Western blot analysis | OPCML is negatively regulated by miR-224-5p in Gastric cancer tissues. |
| hsa-mir-193a | Carcinoma, Hepatocellular | Target | 30710422 | qRT-PCR; Dual luciferase reporter assay; RNA pull-down assay | miR-193a-5p inhibits the growth of hepatocellular carcinoma by targeting SPOCK1. |
| hsa-mir-31 | Gastric Neoplasms | Target | 30677405 | Silico analysis; Dual luciferase reporter assay | Zeste homolog 2 (ZH2) is the potential target of miR-31 in AGS cells to inhibit Gastric cancer. |
| hsa-mir-218-1 | Carcinoma, Hepatocellular | Target | 30003726 | Fluorescence protein analysis; RT-qPCR; Western blotting | miR-218 suppresses the growth of hepatocellular carcinoma by inhibiting the expression of proto-oncogene Bmi-1. |
| hsa-mir-148a | Prostate Neoplasms | Tissue | 20406806 | the trypan blue; dye exclusion assay | miR-148a expression levels are lower in PC3 and DU145 hormone-refractory prostate cancer cells than PrEC normal human prostate epithelial cells. |
| hsa-mir-218-1 | Breast Neoplasms | Target | 29378184 | RT-qPCR analysis; Luciferase reporter assay; Cancer biostatistical analysis | miR-218 regulates breast cancer progression by targeting Lamins. |
| hsa-let-7 | Carcinoma, Hepatocellular | Target | 27821157 | MTT assay; western blot; immunofluorescence; luciferase-reporter assay | Let-7 inhibits the self-renewal of stem cell-like cells by regulating Wnt signaling pathwayand EMT. |
| hsa-mir-193a | Colorectal Carcinoma | Target | 29104111 | qRT-PCR; Western bolt analysis | MiR-193a-3p plays a tumor suppressive role by targeting KRAS in colorectal adenocarcinoma patients. |
| hsa-mir-200c | Carcinoma, Cervical | Target | 27693631 | Luciferase reporter; qRT-PCR assays | Disrupting MALAT1/miR-200c sponge decreases invasion and migration in endometrioid endometrial carcinoma. |
| hsa-mir-34c | Ovarian Neoplasms | Target | 32308421 | qRT-PCR; MTT; Western blot assays; Immunoprecipitation; Flow cytometry analysis | miR-34c targets MET to improve the Anti-Tumor effect of Cisplatin on ovarian cancer. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, D.-L.; Yu, Z.-G.; Han, G.-S.; Li, J.; Anh, V. Heterogeneous Types of miRNA-Disease Associations Stratified by Multi-Layer Network Embedding and Prediction. Biomedicines 2021, 9, 1152. https://doi.org/10.3390/biomedicines9091152
Yu D-L, Yu Z-G, Han G-S, Li J, Anh V. Heterogeneous Types of miRNA-Disease Associations Stratified by Multi-Layer Network Embedding and Prediction. Biomedicines. 2021; 9(9):1152. https://doi.org/10.3390/biomedicines9091152
Chicago/Turabian StyleYu, Dong-Ling, Zu-Guo Yu, Guo-Sheng Han, Jinyan Li, and Vo Anh. 2021. "Heterogeneous Types of miRNA-Disease Associations Stratified by Multi-Layer Network Embedding and Prediction" Biomedicines 9, no. 9: 1152. https://doi.org/10.3390/biomedicines9091152
APA StyleYu, D.-L., Yu, Z.-G., Han, G.-S., Li, J., & Anh, V. (2021). Heterogeneous Types of miRNA-Disease Associations Stratified by Multi-Layer Network Embedding and Prediction. Biomedicines, 9(9), 1152. https://doi.org/10.3390/biomedicines9091152