
A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure


Abstract
:1. Introduction
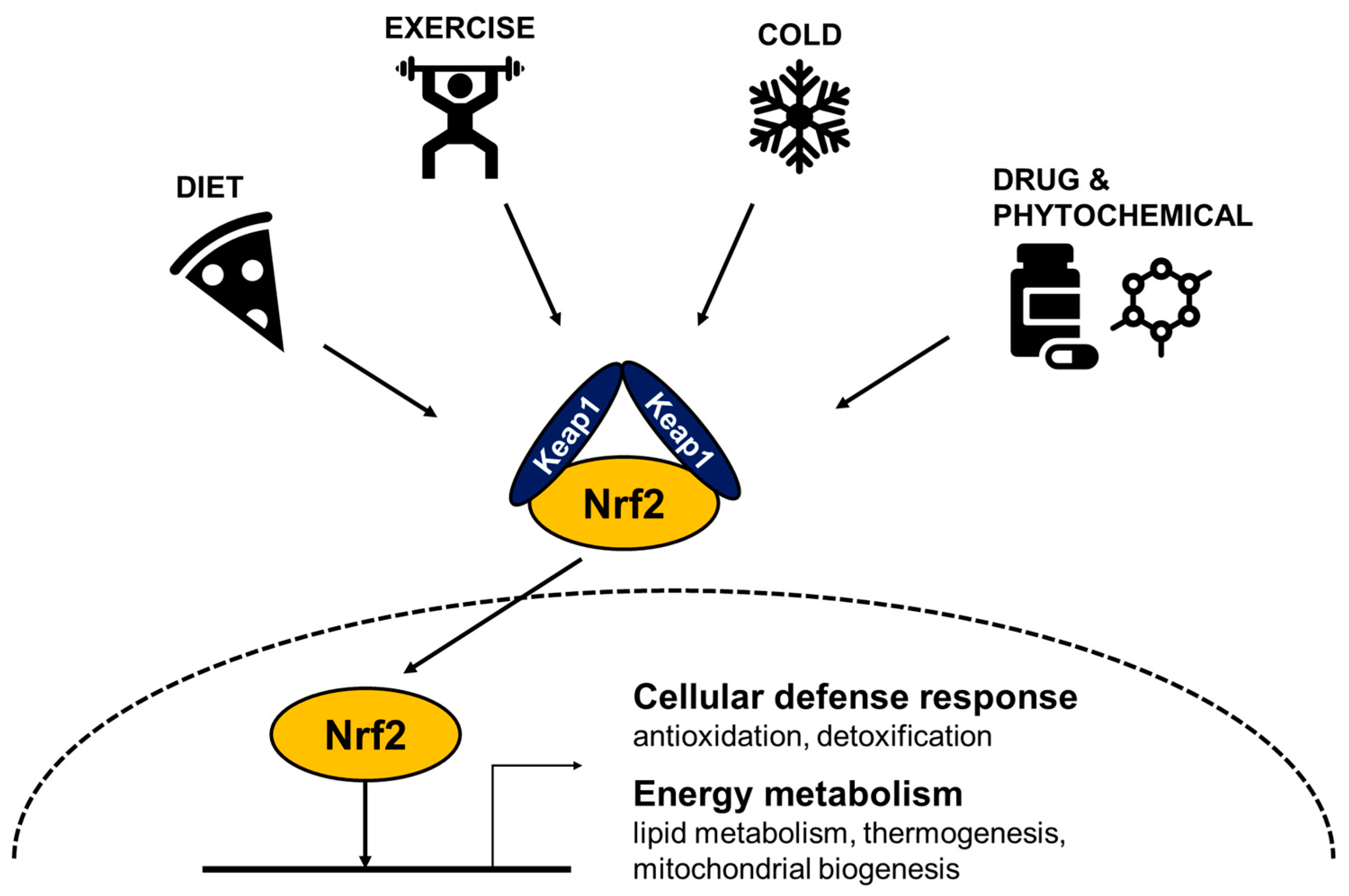
2. Stress Inducible Nrf2 Responds to Cellular Stress
2.1. Oxidative Stress
2.2. Xenobiotic, Phytochemical and Electrophilic Stress
2.3. Metabolic Stress
3. Mitochondrial ROS Signaling Induces Thermogenesis
4. The Role of Nrf2 in Energy Expenditure
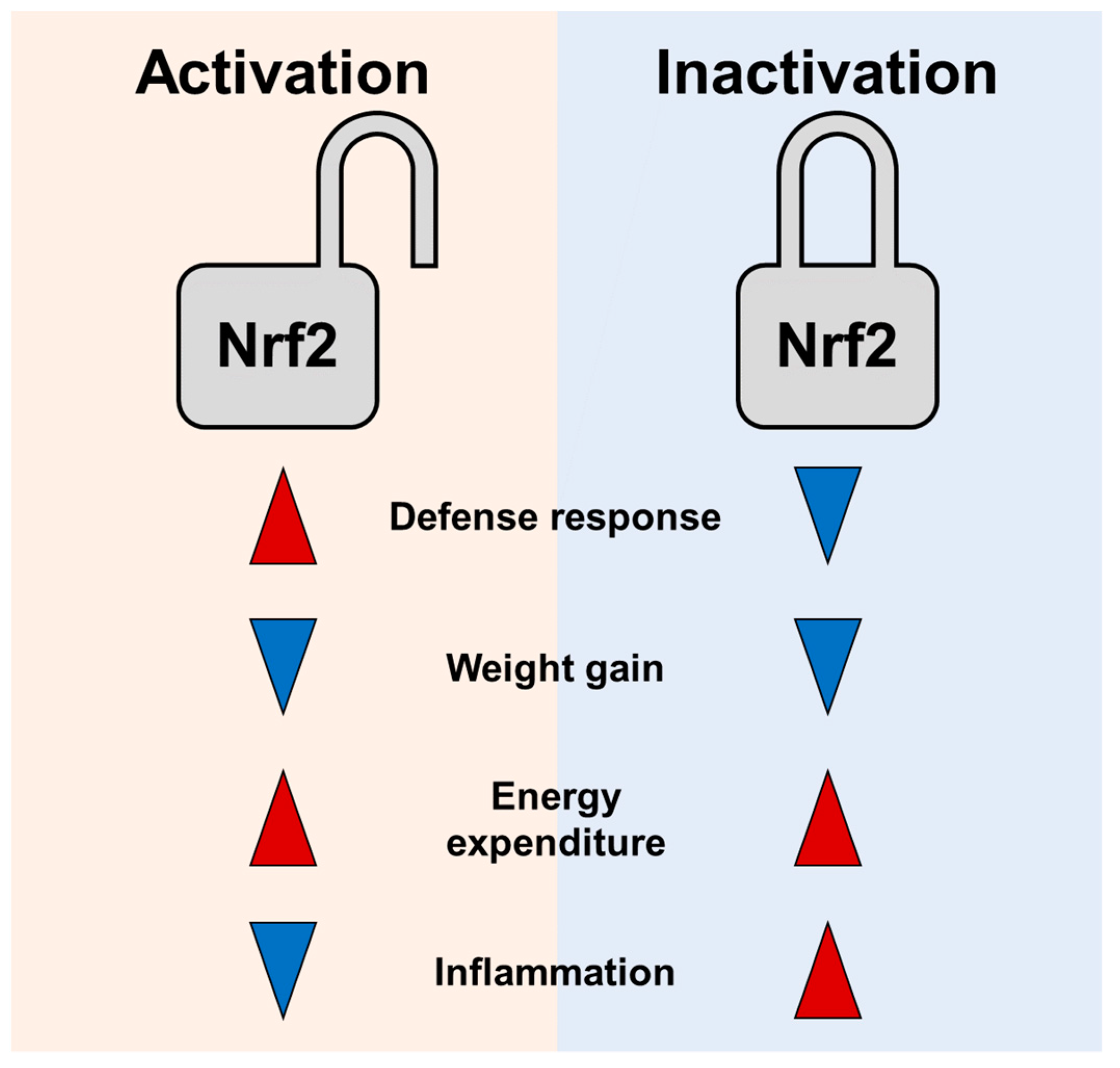
4.1. Nrf2 Inhibition Increases Thermogenesis and Energy Expenditure
4.2. Nrf2 Activation Stimulates Energy Metabolism and Prevents Obesity
5. How to Resolve Similar Effects of Nrf2 Activation and Inhibition in Preventing Obesity?
6. Nrf2 Activation as Strategies to Enhance Energy Expenditure in Obese Conditions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Crompton, A.W.; Taylor, C.R.; Jagger, J.A. Evolution of homeothermy in mammals. Nature 1978, 272, 333–336. [Google Scholar] [CrossRef]
- Betz, M.J.; Enerbäck, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef]
- Chang, S.-H.; Song, N.-J.; Choi, J.H.; Yun, U.J.; Park, K.W. Mechanisms underlying UCP1 dependent and independent adipocyte thermogenesis. Obes. Rev. 2019, 20, 241–251. [Google Scholar] [CrossRef]
- Foster, D.O.; Frydman, M.L. Brown Adipose Tissue: The Dominant Site of Nonshivering Thermogenesis in the Rat. Galanin 1978, 32, 147–151. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Brand, M. The Regulation and Physiology of Mitochondrial Proton Leak. Physiology 2011, 26, 192–205. [Google Scholar] [CrossRef]
- Saito, M. Human brown adipose tissue: Regulation and anti-obesity potential. Endocr. J. 2014, 61, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, A.; Relat, J.; Vidal-Puig, A. Pharmacological strategies for targeting BAT thermogenesis. Trends Pharmacol. Sci. 2013, 34, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem. 2017, 292, 16810–16816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Adjeitey, C.N.; Xuan, J.Y.; Harper, M. Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J. 2012, 26, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Stier, A.; Bize, P.; Habold, C.; Bouillaud, F.; Massemin, S.; Criscuolo, F. Mitochondrial uncoupling prevents cold-induced oxidative stress: A case study using UCP1 knock-out mice. J. Exp. Biol. 2013, 217 Pt 4, 624–630. [Google Scholar] [CrossRef] [Green Version]
- De Quiroga, G.B.; Lopez-Torres, M.; Pérez-Campo, R.; Abelenda, M.; Nava, M.P.; Puerta, M.L. Effect of cold acclimation on GSH, antioxidant enzymes and lipid peroxidation in brown adipose tissue. Biochem. J. 1991, 277, 289–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-Imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin Ameliorates Obesity and Insulin Resistance Through Adipose Tissue Browning and Reduction of Metabolic Endotoxemia in Mice. Diabetes 2017, 66, 1222–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Chang, S.-H.; Yang, D.K.; Song, N.-J.; Yun, U.J.; Park, K.W.; Bell, A.; Chiappe, L.M. Sesamol Increases Ucp1 Expression in White Adipose Tissues and Stimulates Energy Expenditure in High-Fat Diet-Fed Obese Mice. Nutrients 2020, 12, 1459. [Google Scholar] [CrossRef]
- Schneider, K.; Valdez, J.; Nguyen, J.; Vawter, M.; Galke, B.; Kurtz, T.W.; Chan, J.Y. Increased Energy Expenditure, Ucp1 Expression, and Resistance to Diet-induced Obesity in Mice Lacking Nuclear Factor-Erythroid-2-related Transcription Factor-2 (Nrf2). J. Biol. Chem. 2016, 291, 7754–7766. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 System Prevents Onset of Diabetes Mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabo-lism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.W.; Saleh, M.; Higginson, D.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; González-Rodríguez, Á.; Gómez-Guerrero, C.; Moreno, J.A. Editorial: Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 10, 1149. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive Oxygen Species in Health and Disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Kumagai, Y.; Akiyama, M.; Unoki, T. Adaptive Responses to Electrophilic Stress and Reactive Sulfur Species as their Regulator Molecules. Toxicol. Res. 2019, 35, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1–Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.A.; Sawa, T.; Akaike, T. Protein cysteine S-guanylation and electrophilic signal transduction by endogenous ni-tro-nucleotides. Amino Acids 2011, 41, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Suzuki, T.; Yamamoto, M. Roles Nrf2 Plays in Myeloid Cells and Related Disorders. Oxidative Med. Cell. Longev. 2013, 2013, 529219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraweera, T.L.; Rupasinghe, H.P.; Dellaire, G.; Xu, Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, L.W. Protective effects of 2(3)-tert-butyl-4-hydroxyanisole on chemical carcinogenesis. Food Chem. Toxicol. 1986, 24, 1099–1102. [Google Scholar] [CrossRef]
- Iverson, F. Phenolic antioxidants: Health protection branch studies on butylated hydroxyanisole. Cancer Lett. 1995, 93, 49–54. [Google Scholar] [CrossRef]
- Otsuki, A.; Yamamoto, M. Cis-element architecture of Nrf2–sMaf heterodimer binding sites and its relation to diseases. Arch. Pharmacal Res. 2020, 43, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.F.; Jacobs, D.R.; Gross, M.D.; Schreiner, P.J.; Williams, O.D.; Lee, D.-H. Longitudinal associations between body mass index and serum carotenoids: The CARDIA study. Br. J. Nutr. 2006, 95, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Wallström, P.; Wirfält, E.; Lahmann, P.H.; Gullberg, B.; Janzon, L.; Berglund, G. Serum concentrations of beta-carotene and alpha-tocopherol are associated with diet, smoking, and general and central adiposity. Am. J. Clin. Nutr. 2001, 73, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.; Britton, J.R.; Leonardi-Bee, J. Association between antioxidant vitamins and asthma outcome measures: Systematic review and meta-analysis. Thorax 2009, 64, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masschelin, P.M.; Cox, A.R.; Chernis, N.; Hartig, S.M. The Impact of Oxidative Stress on Adipose Tissue Energy Balance. Front. Physiol. 2020, 10, 1638. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.; Thompson, C.B. Cellular Metabolic Stress: Considering How Cells Respond to Nutrient Excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Lettieri-Barbato, D. Redox control of non-shivering thermogenesis. Mol. Metab. 2019, 25, 11–19. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.E.; Vale, C.; Walker, M.; Alberti, K.G.; Mathers, J.C. Dietary carbohydrates and insulin sensitivity: A review of the evidence and clinical implications. Am. J. Clin. Nutr. 1997, 66, 1072–1085. [Google Scholar] [CrossRef] [PubMed]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Done, A.J.; Traustadóttir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute exercise induced mitochondrial H2O2 production in mouse skeletal muscle: Association with p(66Shc) and FOXO3a signaling and antioxidant enzymes. Oxid. Med. Cell. Longev. 2015, 2015, 536456. [Google Scholar] [CrossRef]
- Zoladz, J.A.; Koziel, A.; Broniarek, I.; Woyda-Płoszczyca, A.M.; Ogrodna, K.; Majerczak, J.; Celichowski, J.; Szkutnik, Z.; Jarmuszkiewicz, W. Effect of temperature on fatty acid metabolism in skeletal muscle mitochondria of untrained and endurance-trained rats. PLoS ONE 2017, 12, e0189456. [Google Scholar] [CrossRef] [Green Version]
- Coleman, V.; Sa-Nguanmoo, P.; Koenig, J.; Schulz, T.J.; Grune, T.; Klaus, S.; Kipp, A.P.; Ost, M. Partial involvement of Nrf2 in skeletal muscle mitohormesis as an adaptive response to mitochondrial uncoupling. Sci. Rep. 2018, 8, 2446. [Google Scholar] [CrossRef] [PubMed]
- Eskla, K.-L.; Porosk, R.; Reimets, R.; Visnapuu, T.; Vasar, E.; Hundahl, C.A.; Luuk, H. Hypothermia augments stress response in mammalian cells. Free. Radic. Biol. Med. 2018, 121, 157–168. [Google Scholar] [CrossRef]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Elía, E.F.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Collins, S. β-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. 2011, 2, 102. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H.; Jang, J.Y.; Oh, S.; Yoon, J.H.; Jo, D.G.; Yun, U.J.; Park, K.W. Nrf2 induces Ucp1 expression in adipocytes in response to beta3-AR stimulation and enhances oxygen consumption in high-fat diet-fed obese mice. BMB Rep. 2021, 54, 419–424. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125 Pt 4, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.; Partridge, L.; Gems, D.; et al. Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nat. Cell Biol. 2002, 415, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Buffolo, M.; Pires, K.M.; Pei, S.; Scherer, P.E.; Boudina, S. Adipocyte-Specific Deletion of Manganese Superoxide Dismutase Protects from Diet-Induced Obesity Through Increased Mitochondrial Uncoupling and Biogenesis. Diabetes 2016, 65, 2639–2651. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Barbato, D.L.; Tatulli, G.; Cannata, S.M.; Bernardini, S.; Aquilano, K.; Ciriolo, M.R. Glutathione Decrement Drives Thermogenic Program in Adipose Cells. Sci. Rep. 2015, 5, 13091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, S.P.; Chouchani, E.T.; Boudina, S. Stress turns on the heat: Regulation of mitochondrial biogenesis and UCP1 by ROS in adipocytes. Adipocyte 2017, 6, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Kendig, E.L.; Chen, Y.; Krishan, M.; Johansson, E.; Schneider, S.N.; Genter, M.B.; Nebert, D.W.; Shertzer, H.G. Lipid metabolism and body composition in Gclm (−/−) mice. Toxicol. Appl. Pharmacol. 2011, 257, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-A.; Lee, I.-K. The Role of Nrf2: Adipocyte Differentiation, Obesity, and Insulin Resistance. Oxid. Med. Cell. Longev. 2013, 2013, 184598. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhou, S.; Jiang, X.; Wang, Y.-H.; Li, F.; Wang, Y.-G.; Zheng, Y.; Cai, L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 16, 35–45. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fu, X.; Liao, X.; Li, Y. Nrf2 Activators as Dietary Phytochemicals Against Oxidative Stress, Inflammation, and Mitochondrial Dysfunction in Autism Spectrum Disorders: A Systematic Review. Front. Psychiatry 2020, 11, 561998. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, Q.; Liu, H.; Song, Z.; Chen, W. Phytochemical compounds targeting on Nrf2 for chemoprevention in colorectal cancer. Eur. J. Pharmacol. 2020, 887, 173588. [Google Scholar] [CrossRef]
- Lu, M.-C.; Ji, J.-A.; Jiang, Z.-Y.; You, Q.-D. The Keap1-Nrf2-ARE Pathway as a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 (corrected)(NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef] [Green Version]
- He, H.-J.; Wang, G.-Y.; Gao, Y.; Ling, W.-H.; Guo-Yu, W.; Jin, T.-R. Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet-fed mice. World J. Diabetes 2012, 3, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Leung, L.; Xue, P.; Wang, W.; Hou, Y.; Liu, D.; Yehuda-Shnaidman, E.; Lee, C.; Lau, J.; Kurtz, T.W.; et al. Deficiency in the Nuclear Factor E2-related Factor-2 Transcription Factor Results in Impaired Adipogenesis and Protects against Diet-induced Obesity. J. Biol. Chem. 2010, 285, 9292–9300. [Google Scholar] [CrossRef] [Green Version]
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose Deficiency of Nrf2 in ob/ob Mice Results in Severe Metabolic Syndrome. Diabetes 2012, 62, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Kulkarni, S.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 Activity Worsens Insulin Resistance, Impairs Lipid Accumulation in Adipose Tissue, and Increases Hepatic Steatosis in Leptin-Deficient Mice. Diabetes 2013, 61, 3208–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- More, V.R.; Xu, J.; Shimpi, P.C.; Belgrave, C.; Luyendyk, J.P.; Yamamoto, M.; Slitt, A.L. Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic. Biol. Med. 2013, 61, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Eguchi, N.; Lau, H.; Ichii, H. The Role of the Nrf2 Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6973. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.; Habeos, I.G. Nrf2 Represses FGF21 During Long-Term High-Fat Diet-Induced Obesity in Mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dasuri, K.; Fernandez-Kim, S.-O.; Bruce-Keller, A.J.; Keller, J. Adipose-specific ablation of Nrf2 transiently delayed high-fat diet-induced obesity by altering glucose, lipid and energy metabolism of male mice. Am. J. Transl. Res. 2016, 8, 5309–5319. [Google Scholar]
- Meakin, P.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.; Dinkova-Kostova, A.; Dillon, J.; Hayes, J.D.; Ashford, M. Susceptibility of Nrf2-Null Mice to Steatohepatitis and Cirrhosis upon Consumption of a High-Fat Diet Is Associated with Oxidative Stress, Perturbation of the Unfolded Protein Response, and Disturbance in the Expression of Metabolic Enzymes but Not with Insulin Resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-K.J.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2012, 264, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Aleksunes, L.M.; Yeager, R.L.; Gyamfi, M.A.; Esterly, N.; Guo, G.L.; Klaassen, C.D. NF-E2-Related Factor 2 Inhibits Lipid Accumulation and Oxidative Stress in Mice Fed a High-Fat Diet. J. Pharmacol. Exp. Ther. 2008, 325, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.; Leibel, R.; Tortoriello, D.V. Dietary Curcumin Significantly Improves Obesity-Associated Inflammation and Diabetes in Mouse Models of Diabesity. Endocrinology 2008, 149, 3549–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Chattopadhyay, A. Nrf2–ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.S.; Chan, J.Y. Emerging Role of Nrf2 in Adipocytes and Adipose Biology. Adv. Nutr. 2013, 4, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88 Pt B, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Morra, V.B.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. (BBA) Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.; Ivanov, D.; Tain, L.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 Signaling Regulates Oxidative Stress Tolerance and Lifespan in Drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakiri, E.; Gumeni, S.; Iliaki, K.K.; Benaki, D.; Vougas, K.; Sykiotis, G.; Gorgoulis, V.; Mikros, E.; Scorrano, L.; Trougakos, I.P. Hyperactivation of Nrf2 increases stress tolerance at the cost of aging acceleration due to metabolic deregulation. Aging Cell 2019, 18, e12845. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.D.P.; Baumeister, R.; Blackwell, T.K. Direct Inhibition of the Longevity-Promoting Factor SKN-1 by Insulin-like Signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Suzuki, T.; Harigae, H.; Romeo, P.-H.; Yamamoto, M.; Motohashi, H. NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell. Biol. 2017, 37, e00086-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Keum, Y.-S. NRF2, a Key Regulator of Antioxidants with Two Faces towards Cancer. Oxid. Med. Cell. Longev. 2016, 2016, 2746457. [Google Scholar] [CrossRef] [Green Version]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Probst, B.L.; McCauley, L.; Trevino, I.; Wigley, W.C.; Ferguson, D.A. Cancer Cell Growth Is Differentially Affected by Constitutive Activation of NRF2 by KEAP1 Deletion and Phar-macological Activation of NRF2 by the Synthetic Triterpenoid, RTA. PLoS ONE 2015, 10, e0135257. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Korga, A.; Ostrowska-Lesko, M.; Jozefczyk, A.; Iwan, M.; Wojcik, R.; Zgorka, G.; Herbet, M.; Vilarrubla, G.G.; Dudka, J. Apigenin and hesperidin augment the toxic effect of doxorubicin against HepG2 cells. BMC Pharmacol. Toxicol. 2019, 20, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabzichi, M.; Hamishehkar, H.; Ramezani, F.; Sharifi, S.; Tabasinezhad, M.; Pirouzpanah, M.; Ghanbari, P.; Samadi, N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5311–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Mouse | Background | Diet (Start/Duration) | Body Mass | IR | Oxidative Stress | Inflammation | EE (Ucp1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Nrf2 KO | C57BL6;129SV | HFD (3–4 w/12 w) | ↓ | [81] | ||||
| Nrf2 KO | C57BL6 | HFD (9–10 w/180 d) | ↓ | ↓ | [87] | |||
| Nrf2 KO | C57BL6 | HFWD (12 w/12 w) | ↔ | ↔ | ↓ (GSH in liver) | [90] | ||
| Nrf2 KO | ob/ob | SD (4 w/11 w) | ↓ | ↑ | ↓ (WAT) | [82] | ||
| Nrf2 KO | C57BL6 | HFD (8 w/4 w) | ↔ | ↑ (MDA in liver) | [91] | |||
| Nrf2 KO | C57BL6 | HFD (8–10 w/16–20 w) | ↓ | ↑ | ↑ (GSSG, MDA in liver) | ↑ (liver) | ↑ | [89] |
| Nrf2 KO | C57BL6 | HFD (12 w/6 w) | ↓ | ↑ | ↓ (GSH/GSSG in WAT) | ↑ (↔RER) (↑in WAT) | [17] | |
| Adipo-Nrf2 KO | C57BL6 | HFD (6 w/14 w) | ↓ | ↓ RER in SD | [88] | |||
| Adipo-Nrf2 KO | Albino C57BL6 | HFD (8 w/170 d) | ↔ | ↑ | ↔ | ↔ (↑iWAT ↓eWAT) | [92] |
| Treatment /Mouse | Background | Diet (Start/Duration) | Body Mass | IR | Oxidative Stress | Inflammation | EE (Ucp1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pharmacological activation | ||||||||
| CDDO-Im | C57BL6 | HFD (6–7 w/95 d) | ↓ | ↑ | [14] | |||
| Glucoraphanin | C57BL6 | HFD (8 w/14 w) | ↓ | ↓ | ↓ (liver, eWAT) | ↑ (↑WAT, ↔BAT) | [15] | |
| Oltipraz | C57BL6 | HFD (5 w/28 w) | ↓ | ↓ | ↑ (GSH/GSSG) | ↓ (eWAT) | [79] | |
| Curcumin | C57BL6 | HFD (9 w/18 w) | ↔ | ↓ | ↓ (muscle) ↔ (adipose and liver) | [80] | ||
| Curcumin | C57BL6 | HFD (3–5 w/15 w) | ↓ | ↓ | ↓ (liver, WAT) | [93] | ||
| Curcumin | ob/ob | SD (8–10 w/14–18 w) | ↓ | ↓ | ↓ (liver, WAT) | [93] | ||
| Sesamol | C57BL6 | HFD (8 w/12 w) | ↓ | ↓ | ↓ (iWAT) | ↑ (↑iWAT) | [16] | |
| Genetic activation | ||||||||
| Keap1 KD | C57BL6 | HFD (9 w/36 d) | ↓ | [83] | ||||
| Keap1 KD | ob/ob | SD (4 w/8 w) | ↔ | ↑ | [83] | |||
| Keap1 KD | C57BL6 | HFWD (12 w/12 w) | ↔ | ↔ | [90] | |||
| Keap1 KD | C57BL6 | HFD (3 w/24 w) | ↑ | ↑ | ↑ (liver, WAT) | ↑ | [84] | |
| Keap1 flox/− | ICR | SD/HCD (4 w/12 w) | ↓ | ↓ | ↑ (↔BAT) | [18] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.-H.; Lee, J.-S.; Yun, U.J.; Park, K.W. A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure. Biomedicines 2021, 9, 1196. https://doi.org/10.3390/biomedicines9091196
Chang S-H, Lee J-S, Yun UJ, Park KW. A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure. Biomedicines. 2021; 9(9):1196. https://doi.org/10.3390/biomedicines9091196
Chicago/Turabian StyleChang, Seo-Hyuk, Jeong-Soo Lee, Ui Jeong Yun, and Kye Won Park. 2021. "A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure" Biomedicines 9, no. 9: 1196. https://doi.org/10.3390/biomedicines9091196
APA StyleChang, S.-H., Lee, J.-S., Yun, U. J., & Park, K. W. (2021). A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure. Biomedicines, 9(9), 1196. https://doi.org/10.3390/biomedicines9091196