
Macrophages in Atherosclerosis, First or Second Row Players?


Abstract
:1. Introduction
1.1. At the Beginning of Atherosclerosis
1.2. Monocytes as Initiators of Atherosclerosis
2. Vascular Smooth Muscle Cells and Early Atherosclerosis
2.1. At the Origin of Foam Cell Formation: Macrophages or VSMCs?
2.2. VSMC Foam Cell Formation
2.3. Scavenger Receptors Expression, a Step towards Foam Cell Formation
2.4. Towards the Necrotic Core Formation
3. “Of Mice and Men”
3.1. The Limits of Experimentations on Animal Models
3.2. From Formation to Destruction of the Fibrous Cap: The Crucial Role of Macrophages
3.3. VSMCs Are Indissociable from Fibrous Cap Formation and Rupture
3.4. Intraplaque Hemorrhage and Neovascularization
3.5. Towards Plaque Calcification
4. Promising Macrophage and VSMC Targeting Therapeutic Strategies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/fr/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 2 March 2020).
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular Smooth Muscle Cells in Atherosclerosis: Time for a Reassessment. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling: Molecular, Cellular, and Vascular Behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reidy, M.A.; Bowyer, D.E. Scanning Electron Microscopy of Arteries. The Morphology of Aortic Endothelium in Haemodynamically Stressed Areas Associated with Branches. Atherosclerosis 1977, 26, 181–194. [Google Scholar] [CrossRef]
- Tarbell, J.M. Shear Stress and the Endothelial Transport Barrier. Cardiovasc. Res. 2010, 87, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial Permeability, LDL Deposition, and Cardiovascular Risk Factors-a Review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease: Pathophysiological, Genetic, and Therapeutic Insights: A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Yu, J.; Suárez, Y.; Rahner, C.; Dávalos, A.; Lasunción, M.A.; Sessa, W.C. Genetic Evidence Supporting a Critical Role of Endothelial Caveolin-1 during the Progression of Atherosclerosis. Cell Metab. 2009, 10, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.M.; Sugiyama, M.G.; Fung, K.Y.Y.; Gao, Y.; Wang, C.; Levy, A.S.; Azizi, P.; Roufaiel, M.; Zhu, S.-N.; Neculai, D.; et al. A Novel Assay Uncovers an Unexpected Role for SR-BI in LDL Transcytosis. Cardiovasc. Res. 2015, 108, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 Drives Endothelial Cell LDL Transcytosis via DOCK4 to Promote Atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-Wide RNAi Screen Reveals ALK1 Mediates LDL Uptake and Transcytosis in Endothelial Cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef]
- Dehouck, B.; Fenart, L.; Dehouck, M.P.; Pierce, A.; Torpier, G.; Cecchelli, R. A New Function for the LDL Receptor: Transcytosis of LDL across the Blood-Brain Barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef]
- Nakashima, Y.; Chen, Y.-X.; Kinukawa, N.; Sueishi, K. Distributions of Diffuse Intimal Thickening in Human Arteries: Preferential Expression in Atherosclerosis-Prone Arteries from an Early Age. Virchows Arch. Int. J. Pathol. 2002, 441, 279–288. [Google Scholar] [CrossRef]
- Iverius, P.H. The Interaction between Human Plasma Lipoproteins and Connective Tissue Glycosaminoglycans. J. Biol. Chem. 1972, 247, 2607–2613. [Google Scholar] [CrossRef]
- Allahverdian, S.; Ortega, C.; Francis, G.A. Smooth Muscle Cell-Proteoglycan-Lipoprotein Interactions as Drivers of Atherosclerosis. Handb. Exp. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Murry, C.E.; Gipaya, C.T.; Bartosek, T.; Benditt, E.P.; Schwartz, S.M. Monoclonality of Smooth Muscle Cells in Human Atherosclerosis. Am. J. Pathol. 1997, 151, 697–705. [Google Scholar]
- Chung, I.M.; Schwartz, S.M.; Murry, C.E. Clonal Architecture of Normal and Atherosclerotic Aorta: Implications for Atherogenesis and Vascular Development. Am. J. Pathol. 1998, 152, 913–923. [Google Scholar]
- Sterpetti, A.V.; Cucina, A.; Morena, A.R.; Di Donna, S.; D’Angelo, L.S.; Cavalarro, A.; Stipa, S. Shear Stress Increases the Release of Interleukin-1 and Interleukin-6 by Aortic Endothelial Cells. Surgery 1993, 114, 911–914. [Google Scholar]
- Huang, R.B.; Gonzalez, A.L.; Eniola-Adefeso, O. Laminar Shear Stress Elicit Distinct Endothelial Cell E-Selectin Expression Pattern via TNFα and IL-1β Activation. Biotechnol. Bioeng. 2013, 110, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylä-Herttuala, S.; Lipton, B.A.; Rosenfeld, M.E.; Särkioja, T.; Yoshimura, T.; Leonard, E.J.; Witztum, J.L.; Steinberg, D. Expression of Monocyte Chemoattractant Protein 1 in Macrophage-Rich Areas of Human and Rabbit Atherosclerotic Lesions. Proc. Natl. Acad. Sci. USA 1991, 88, 5252–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, U.; Henning, S.; Wenschuh, H.; Mayer, K.; Seeger, W.; Lohmeyer, J. Role of Endothelial MCP-1 in Monocyte Adhesion to Inflamed Human Endothelium under Physiological Flow. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2584–H2591. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Yoshida, H.; Satoh, K. Regulation of CX3CL1/Fractalkine Expression in Endothelial Cells. J. Atheroscler. Thromb. 2004, 11, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, D.C.; Varner, S.E.; Nerem, R.M.; Medford, R.M.; Alexander, R.W. Oscillatory Shear Stress Stimulates Adhesion Molecule Expression in Cultured Human Endothelium. Circ. Res. 1998, 82, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Olivares, R.; Ducimetière, P.; Claude, J.R. Monocyte Count: A Risk Factor for Coronary Heart Disease? Am. J. Epidemiol. 1993, 137, 49–53. [Google Scholar] [CrossRef]
- Nielsen, L.B. Transfer of Low Density Lipoprotein into the Arterial Wall and Risk of Atherosclerosis. Atherosclerosis 1996, 123, 1–15. [Google Scholar] [CrossRef]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi Monocytes Dominate Hypercholesterolemia-Associated Monocytosis and Give Rise to Macrophages in Atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Cookson, F.B. The Origin of Foam Cells in Atherosclerosis. Br. J. Exp. Pathol. 1971, 52, 62–69. [Google Scholar]
- Gerrity, R.G. The Role of the Monocyte in Atherogenesis: II. Migration of Foam Cells from Atherosclerotic Lesions. Am. J. Pathol. 1981, 103, 191–200. [Google Scholar]
- Taylor, R.G.; Lewis, J.C. Endothelial Cell Proliferation and Monocyte Adhesion to Atherosclerotic Lesions of White Carneau Pigeons. Am. J. Pathol. 1986, 125, 152–160. [Google Scholar]
- Tolani, S.; Pagler, T.A.; Murphy, A.J.; Bochem, A.E.; Abramowicz, S.; Welch, C.; Nagareddy, P.R.; Holleran, S.; Hovingh, G.K.; Kuivenhoven, J.A.; et al. Hypercholesterolemia and Reduced HDL-C Promote Hematopoietic Stem Cell Proliferation and Monocytosis: Studies in Mice and FH Children. Atherosclerosis 2013, 229, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, Inflammation and Innate Immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective Cholesterol Metabolism in Haematopoietic Stem Cells Promotes Monocyte-Driven Atherosclerosis in Rheumatoid Arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgendorf, I.; Swirski, F.K.; Robbins, C.S. Monocyte Fate in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Cappellari, R.; D’Anna, M.; Bonora, B.M.; Rigato, M.; Cignarella, A.; Avogaro, A.; Fadini, G.P. Shift of Monocyte Subsets along Their Continuum Predicts Cardiovascular Outcomes. Atherosclerosis 2017, 266, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Imhof, B.A.; Jemelin, S.; Ballet, R.; Vesin, C.; Schapira, M.; Karaca, M.; Emre, Y. CCN1/CYR61-Mediated Meticulous Patrolling by Ly6Clow Monocytes Fuels Vascular Inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, E4847–E4856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages towards a Reparative Phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Lessard, A.-J.; LeBel, M.; Egarnes, B.; Préfontaine, P.; Thériault, P.; Droit, A.; Brunet, A.; Rivest, S.; Gosselin, J. Triggering of NOD2 Receptor Converts Inflammatory Ly6Chigh into Ly6Clow Monocytes with Patrolling Properties. Cell Rep. 2017, 20, 1830–1843. [Google Scholar] [CrossRef] [Green Version]
- Polletti, S.; Natoli, G. Understanding Spontaneous Conversion: The Case of the Ly6C- Monocyte. Immunity 2017, 46, 764–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils Promote the Development of Reparative Macrophages Mediated by ROS to Orchestrate Liver Repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.; Vengrenyuk, Y.; Ramsey, S.A.; Vila, N.R.; Girgis, N.M.; Liu, J.; Gusarova, V.; Gromada, J.; Weinstock, A.; Moore, K.J.; et al. Inflammatory Ly6Chi Monocytes and Their Conversion to M2 Macrophages Drive Atherosclerosis Regression. J. Clin. Investig. 2017, 127, 2904–2915. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Green, S.R.; Han, K.H.; Chen, Y.; Almazan, F.; Charo, I.F.; Miller, Y.I.; Quehenberger, O. The CC Chemokine MCP-1 Stimulates Surface Expression of CX3CR1 and Enhances the Adhesion of Monocytes to Fractalkine/CX3CL1 via P38 MAPK. J. Immunol. Baltim. Md 1950 2006, 176, 7412–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbina, N.V.; Pamer, E.G. Monocyte Emigration from Bone Marrow during Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Stein, O.; Dabach, Y.; Ben-Naim, M.; Halperin, G.; Charo, I.F.; Stein, Y. In CCR2-/- Mice Monocyte Recruitment and Egress of LDL Cholesterol in Vivo Is Impaired. Biochem. Biophys. Res. Commun. 2003, 300, 477–481. [Google Scholar] [CrossRef]
- Landsman, L.; Bar-On, L.; Zernecke, A.; Kim, K.-W.; Krauthgamer, R.; Shagdarsuren, E.; Lira, S.A.; Weissman, I.L.; Weber, C.; Jung, S. CX3CR1 Is Required for Monocyte Homeostasis and Atherogenesis by Promoting Cell Survival. Blood 2009, 113, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Aiello, R.J.; Bourassa, P.A.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte Chemoattractant Protein-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined Inhibition of CCL2, CX3CR1, and CCR5 Abrogates Ly6C(Hi) and Ly6C(Lo) Monocytosis and Almost Abolishes Atherosclerosis in Hypercholesterolemic Mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of Monocyte Chemoattractant Protein-1 Reduces Atherosclerosis in Low Density Lipoprotein Receptor-Deficient Mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Ramirez-Ortiz, Z.G.; Prasad, A.; Griffith, J.W.; Pendergraft, W.F.; Cowley, G.S.; Root, D.E.; Tai, M.; Luster, A.D.; El Khoury, J.; Hacohen, N.; et al. The Receptor TREML4 Amplifies TLR7-Mediated Signaling during Antiviral Responses and Autoimmunity. Nat. Immunol. 2015, 16, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Silbiger, V.N.; Luchessi, A.D.; Hirata, R.D.C.; Lima-Neto, L.G.; Cavichioli, D.; Carracedo, A.; Brión, M.; Dopazo, J.; García-García, F.; dos Santos, E.S.; et al. Novel Genes Detected by Transcriptional Profiling from Whole-Blood Cells in Patients with Early Onset of Acute Coronary Syndrome. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 421, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.K.; Boelte, K.C.; Barb, J.J.; Joehanes, R.; Zhao, X.; Cheng, Q.; Adams, L.; Teer, J.K.; Accame, D.S.; Chowdhury, S.; et al. Integrative DNA, RNA, and Protein Evidence Connects TREML4 to Coronary Artery Calcification. Am. J. Hum. Genet. 2014, 95, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cotto, M.; Guo, L.; Karwan, M.; Sen, S.K.; Barb, J.; Collado, C.J.; Elloumi, F.; Palmieri, E.M.; Boelte, K.; Kolodgie, F.D.; et al. TREML4 Promotes Inflammatory Programs in Human and Murine Macrophages and Alters Atherosclerosis Lesion Composition in the Apolipoprotein E Deficient Mouse. Front. Immunol. 2020, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Xia, L. P-Selectin Glycoprotein Ligand-1 Plays a Crucial Role in the Selective Recruitment of Leukocytes into the Atherosclerotic Arterial Wall. Trends Cardiovasc. Med. 2009, 19, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Ronald, J.A.; Ionescu, C.V.; Rogers, K.A.; Sandig, M. Differential Regulation of Transendothelial Migration of THP-1 Cells by ICAM-1/LFA-1 and VCAM-1/VLA-4. J. Leukoc. Biol. 2001, 70, 601–609. [Google Scholar]
- Yusuf-Makagiansar, H.; Anderson, M.E.; Yakovleva, T.V.; Murray, J.S.; Siahaan, T.J. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a Therapeutic Approach to Inflammation and Autoimmune Diseases. Med. Res. Rev. 2002, 22, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Deng, X.; Chen, L.; Yang, X.; Yu, C. ST6GAL1 Negatively Regulates Monocyte Transendothelial Migration and Atherosclerosis Development. Biochem. Biophys. Res. Commun. 2018, 500, 249–255. [Google Scholar] [CrossRef]
- Fatigati, V.; Murphy, R.A. Actin and Tropomyosin Variants in Smooth Muscles. Dependence on Tissue Type. J. Biol. Chem. 1984, 259, 14383–14388. [Google Scholar] [CrossRef]
- Kargacin, G.J.; Cooke, P.H.; Abramson, S.B.; Fay, F.S. Periodic Organization of the Contractile Apparatus in Smooth Muscle Revealed by the Motion of Dense Bodies in Single Cells. J. Cell Biol. 1989, 108, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Rensen, S.S.M.; Doevendans, P.A.F.M.; van Eys, G.J.J.M. Regulation and Characteristics of Vascular Smooth Muscle Cell Phenotypic Diversity. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2007, 15, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosse, P.R.; Campbell, G.R.; Wang, Z.L.; Campbell, J.H. Smooth Muscle Phenotypic Expression in Human Carotid Arteries. I. Comparison of Cells from Diffuse Intimal Thickenings Adjacent to Atheromatous Plaques with Those of the Media. Lab. Investig. J. Tech. Methods Pathol. 1985, 53, 556–562. [Google Scholar]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A Definition of Initial, Fatty Streak, and Intermediate Lesions of Atherosclerosis. A Report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikari, Y.; McManus, B.M.; Kenyon, J.; Schwartz, S.M. Neonatal Intima Formation in the Human Coronary Artery. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2036–2040. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Chang, P.S.; Wang, Z.; Sutherland, L.; Richardson, J.A.; Small, E.; Krieg, P.A.; Olson, E.N. Activation of Cardiac Gene Expression by Myocardin, a Transcriptional Cofactor for Serum Response Factor. Cell 2001, 105, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Pipes, G.C.T.; Creemers, E.E.; Olson, E.N. The Myocardin Family of Transcriptional Coactivators: Versatile Regulators of Cell Growth, Migration, and Myogenesis. Genes Dev. 2006, 20, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Collagen-Synthesizing Cells in Initial and Advanced Atherosclerotic Lesions of Human Aorta. Atherosclerosis 1997, 130, 133–142. [Google Scholar] [CrossRef]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like Factor 4 Abrogates Myocardin-Induced Activation of Smooth Muscle Gene Expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef] [Green Version]
- Pidkovka, N.A.; Cherepanova, O.A.; Yoshida, T.; Alexander, M.R.; Deaton, R.A.; Thomas, J.A.; Leitinger, N.; Owens, G.K. Oxidized Phospholipids Induce Phenotypic Switching of Vascular Smooth Muscle Cells in Vivo and in Vitro. Circ. Res. 2007, 101, 792–801. [Google Scholar] [CrossRef]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.R.; Murgai, M.; Moehle, C.W.; Owens, G.K. Interleukin-1β Modulates Smooth Muscle Cell Phenotype to a Distinct Inflammatory State Relative to PDGF-DD via NF-ΚB-Dependent Mechanisms. Physiol. Genomics 2012, 44, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudijanto, A. The Role of Vascular Smooth Muscle Cells on the Pathogenesis of Atherosclerosis. Acta Medica Indones. 2007, 39, 86–93. [Google Scholar]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.-L. Smooth Muscle Cell Fate and Plasticity in Atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, A.; Kwartler, C.S.; Kaw, K.; Li, Y.; Kaw, A.; Chen, J.; LeMaire, S.A.; Shen, Y.H.; Milewicz, D.M. Cholesterol-Induced Phenotypic Modulation of Smooth Muscle Cells to Macrophage/Fibroblast-like Cells Is Driven by an Unfolded Protein Response. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Walker-Caprioglio, H.M.; Hunter, D.D.; McGuire, P.G.; Little, S.A.; McGuffee, L.J. Composition in Situ and in Vitro of Vascular Smooth Muscle Laminin in the Rat. Cell Tissue Res. 1995, 281, 187–196. [Google Scholar] [CrossRef]
- Thyberg, J.; Blomgren, K.; Roy, J.; Tran, P.K.; Hedin, U. Phenotypic Modulation of Smooth Muscle Cells after Arterial Injury Is Associated with Changes in the Distribution of Laminin and Fibronectin. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1997, 45, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.J.; Johnson, J.L. Extracellular Matrix and Smooth Muscle Cells. In Inflammation and Atherosclerosis; Wick, G., Grundtman, C., Eds.; Springer: Vienna, Austria, 2012; pp. 435–460. ISBN 978-3-7091-0337-1. [Google Scholar]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial Retention of Atherogenic Lipoproteins in Early Atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Colony-Stimulating Factors in Inflammation and Autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef]
- Fuhrman, B.; Partoush, A.; Volkova, N.; Aviram, M. Ox-LDL Induces Monocyte-to-Macrophage Differentiation in Vivo: Possible Role for the Macrophage Colony Stimulating Factor Receptor (M-CSF-R). Atherosclerosis 2008, 196, 598–607. [Google Scholar] [CrossRef]
- Domschke, G.; Gleissner, C.A. CXCL4-Induced Macrophages in Human Atherosclerosis. Cytokine 2019, 122, 154141. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage Phenotypes in Atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Dolfi, B.; Gallerand, A.; Haschemi, A.; Guinamard, R.R.; Ivanov, S. Macrophage Metabolic Regulation in Atherosclerotic Plaque. Atherosclerosis 2021, 334, 1–8. [Google Scholar] [CrossRef]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, S.; Zamora, D.; Ullevig, S.; Asmis, R. Bioenergetic Profiles Diverge during Macrophage Polarization: Implications for the Interpretation of 18F-FDG PET Imaging of Atherosclerosis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in Transcriptome of Macrophages in Atherosclerosis. J. Cell. Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 Macrophage Polarization by the Crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhika, A.; Jacob, S.S.; Sudhakaran, P.R. Influence of Oxidatively Modified LDL on Monocyte-Macrophage Differentiation. Mol. Cell. Biochem. 2007, 305, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Pryma, C.S.; Ortega, C.; Dubland, J.A.; Francis, G.A. Pathways of Smooth Muscle Foam Cell Formation in Atherosclerosis. Curr. Opin. Lipidol. 2019, 30, 117–124. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Gabbiani, G.; Hansson, G.K. Expression of Class II Transplantation Antigen on Vascular Smooth Muscle Cells in Human Atherosclerosis. J. Clin. Investig. 1985, 76, 125–131. [Google Scholar] [CrossRef]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial Smooth Muscle Cells of Human Aorta Express Macrophage Antigen in Situ and in Vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef]
- Gomez, D.; Shankman, L.S.; Nguyen, A.T.; Owens, G.K. Detection of Histone Modifications at Specific Gene Loci in Single Cells in Histological Sections. Nat. Methods 2013, 10, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-like Cells during Atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-Dependent Phenotypic Modulation of Smooth Muscle Cells Has a Key Role in Atherosclerotic Plaque Pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarrán-Juárez, J.; Kaur, H.; Grimm, M.; Offermanns, S.; Wettschureck, N. Lineage Tracing of Cells Involved in Atherosclerosis. Atherosclerosis 2016, 251, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Gomez, D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1715–1723. [Google Scholar] [CrossRef]
- Nakashima, Y.; Fujii, H.; Sumiyoshi, S.; Wight, T.N.; Sueishi, K. Early Human Atherosclerosis: Accumulation of Lipid and Proteoglycans in Intimal Thickenings Followed by Macrophage Infiltration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Dubland, J.A.; Allahverdian, S.; Besler, K.J.; Ortega, C.; Wang, Y.; Pryma, C.S.; Boukais, K.; Chan, T.; Seidman, M.A.; Francis, G.A. Low LAL (Lysosomal Acid Lipase) Expression by Smooth Muscle Cells Relative to Macrophages as a Mechanism for Arterial Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2021, ATVBAHA120316063. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Xue, C.; Feng, K.; Liu, L.; Zeng, P.; Wang, X.; Chen, Y.; Li, L.; Zhang, Z.; et al. TL1A Inhibits Atherosclerosis in ApoE-Deficient Mice by Regulating the Phenotype of Vascular Smooth Muscle Cells. J. Biol. Chem. 2020, 295, 16314–16327. [Google Scholar] [CrossRef]
- Ismail, N.A.; Alavi, M.Z.; Moore, S. Lipoprotein-Proteoglycan Complexes from Injured Rabbit Aortas Accelerate Lipoprotein Uptake by Arterial Smooth Muscle Cells. Atherosclerosis 1994, 105, 79–87. [Google Scholar] [CrossRef]
- Oörni, K.; Posio, P.; Ala-Korpela, M.; Jauhiainen, M.; Kovanen, P.T. Sphingomyelinase Induces Aggregation and Fusion of Small Very Low-Density Lipoprotein and Intermediate-Density Lipoprotein Particles and Increases Their Retention to Human Arterial Proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1678–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argmann, C.A.; Sawyez, C.G.; Li, S.; Nong, Z.; Hegele, R.A.; Pickering, J.G.; Huff, M.W. Human Smooth Muscle Cell Subpopulations Differentially Accumulate Cholesteryl Ester When Exposed to Native and Oxidized Lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1290–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klouche, M.; Rose-John, S.; Schmiedt, W.; Bhakdi, S. Enzymatically Degraded, Nonoxidized LDL Induces Human Vascular Smooth Muscle Cell Activation, Foam Cell Transformation, and Proliferation. Circulation 2000, 101, 1799–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellan, B.; Rojas, E.; Zhang, C.; Hofmann Bowman, M.A. Enzyme-Modified Non-Oxidized LDL (ELDL) Induces Human Coronary Artery Smooth Muscle Cell Transformation to a Migratory and Osteoblast-like Phenotype. Sci. Rep. 2018, 8, 11954. [Google Scholar] [CrossRef]
- Vijayagopal, P.; Glancy, D.L. Macrophages Stimulate Cholesteryl Ester Accumulation in Cocultured Smooth Muscle Cells Incubated with Lipoprotein-Proteoglycan Complex. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1112–1121. [Google Scholar] [CrossRef]
- Weinert, S.; Poitz, D.M.; Auffermann-Gretzinger, S.; Eger, L.; Herold, J.; Medunjanin, S.; Schmeisser, A.; Strasser, R.H.; Braun-Dullaeus, R.C. The Lysosomal Transfer of LDL/Cholesterol from Macrophages into Vascular Smooth Muscle Cells Induces Their Phenotypic Alteration. Cardiovasc. Res. 2013, 97, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Wang, X.; Zhao, M.; Cai, T.; Liu, P.; Li, J.; Willard, B.; Zu, L.; Zhou, E.; Li, Y.; et al. Macrophage Foam Cell-Derived Extracellular Vesicles Promote Vascular Smooth Muscle Cell Migration and Adhesion. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Wolfbauer, G.; Glick, J.M.; Minor, L.K.; Rothblat, G.H. Development of the Smooth Muscle Foam Cell: Uptake of Macrophage Lipid Inclusions. Proc. Natl. Acad. Sci. USA 1986, 83, 7760–7764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Jiang, H.; Song, W.; Riezman, H.; Tontonoz, P.; Weston, T.A.; Guagliardo, P.; Kim, P.H.; Jung, R.; Heizer, P.; et al. Cultured Macrophages Transfer Surplus Cholesterol into Adjacent Cells in the Absence of Serum or High-Density Lipoproteins. Proc. Natl. Acad. Sci. USA 2020, 117, 10476–10483. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; D’Armiento, F.P.; Mancini, F.P.; Postiglione, A.; Witztum, J.L.; Palumbo, G.; Palinski, W. Fatty Streak Formation Occurs in Human Fetal Aortas and Is Greatly Enhanced by Maternal Hypercholesterolemia. Intimal Accumulation of Low Density Lipoprotein and Its Oxidation Precede Monocyte Recruitment into Early Atherosclerotic Lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Basu, S.K.; Falck, J.R.; Ho, Y.K.; Goldstein, J.L. The Scavenger Cell Pathway for Lipoprotein Degradation: Specificity of the Binding Site That Mediates the Uptake of Negatively-Charged LDL by Macrophages. J. Supramol. Struct. 1980, 13, 67–81. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A Receptor-Mediated Pathway for Cholesterol Homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma Promotes Monocyte/Macrophage Differentiation and Uptake of Oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Diaz, S.; Higa, H.H.; Hayes, B.K.; Varki, A. O-Acetylation and de-O-Acetylation of Sialic Acids. 7- and 9-o-Acetylation of Alpha 2,6-Linked Sialic Acids on Endogenous N-Linked Glycans in Rat Liver Golgi Vesicles. J. Biol. Chem. 1989, 264, 19416–19426. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Oiknine, J.; Aviram, M. Increased Susceptibility to Activation and Increased Uptake of Low Density Lipoprotein by Cholesterol-Loaded Macrophages. Arterioscler. Thromb. J. Vasc. Biol. 1992, 12, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-H.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein Accumulation in Macrophages via Toll-like Receptor-4-Dependent Fluid Phase Uptake. Circ. Res. 2009, 104, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.-N.; Zhou, X.; Lu, Y.-R.; Li, K.; Gao, S.; Yu, C.-Q.; Cui, Y.-L. Dan-Lou Prescription Inhibits Foam Cell Formation Induced by Ox-LDL via the TLR4/NF-ΚB and PPARγ Signaling Pathways. Front. Physiol. 2018, 9, 590. [Google Scholar] [CrossRef]
- Di Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and Pathophysiology of OxLDL Uptake by Vascular Wall Cells in Atherosclerosis. Vascul. Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N.; Bobryshev, Y.V. How Do Macrophages Sense Modified Low-Density Lipoproteins? Int. J. Cardiol. 2017, 230, 232–240. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Pan, Y.; Huang, Z.; Jia, Y.; Zhao, X.; Chen, Y.; Diao, J.; Wan, Q.; Cui, X. Visfatin Induces Cholesterol Accumulation in Macrophages through Up-Regulation of Scavenger Receptor-A and CD36. Cell Stress Chaperones 2013, 18, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Ben, J.; Zhu, X.; Zhang, H.; Chen, Q. Class A1 Scavenger Receptors in Cardiovascular Diseases. Br. J. Pharmacol. 2015, 172, 5523–5530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreurs, M.P.H.; Hubel, C.A.; Bernstein, I.M.; Jeyabalan, A.; Cipolla, M.J. Increased Oxidized Low-Density Lipoprotein Causes Blood-Brain Barrier Disruption in Early-Onset Preeclampsia through LOX-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediators Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ding, Z.; Lin, J.; Guo, Z.; Mehta, J.L. LOX-1 in Macrophage Migration in Response to Ox-LDL and the Involvement of Calpains. Biochem. Biophys. Res. Commun. 2015, 467, 135–139. [Google Scholar] [CrossRef]
- Nan, J.; Xing, Y.-F.; Hu, B.; Tang, J.-X.; Dong, H.-M.; He, Y.-M.; Ruan, D.-Y.; Ye, Q.-J.; Cai, J.-R.; Ma, X.-K.; et al. Endoplasmic Reticulum Stress Induced LOX-1+ CD15+ Polymorphonuclear Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma. Immunology 2018, 154, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, M.; Mackman, N. LPS Induction of Gene Expression in Human Monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated Fatty Acids Trigger TLR4-Mediated Inflammatory Response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell Surface Expression of Mouse Macrosialin and Human CD68 and Their Role as Macrophage Receptors for Oxidized Low Density Lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanzada, A.; Malik, I.A.; Blaschke, M.; Khan, S.; Rahman, H.; Ramadori, G.; Moriconi, F. Identification of CD68(+) Neutrophil Granulocytes in in Vitro Model of Acute Inflammation and Inflammatory Bowel Disease. Int. J. Clin. Exp. Pathol. 2013, 6, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Duan, H.; Qian, Y.; Feng, L.; Wu, Z.; Wang, F.; Feng, J.; Yang, D.; Qin, Z.; Yan, X. Macrophagic CD146 Promotes Foam Cell Formation and Retention during Atherosclerosis. Cell Res. 2017, 27, 352–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Zhao, S.; Braunstein, Z.; Mao, H.; Razavi, M.; Duan, L.; Wei, Y.; Toomey, A.C.; Rajagopalan, S.; Zhong, J. Oxidized LDL Upregulates Macrophage DPP4 Expression via TLR4/TRIF/CD36 Pathways. EBioMedicine 2019, 41, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Nicholson, A.C. Lipoproteins Modulate Expression of the Macrophage Scavenger Receptor. Am. J. Pathol. 1998, 152, 1647–1654. [Google Scholar]
- Dai, Y.; Su, W.; Ding, Z.; Wang, X.; Mercanti, F.; Chen, M.; Raina, S.; Mehta, J.L. Regulation of MSR-1 and CD36 in Macrophages by LOX-1 Mediated through PPAR-γ. Biochem. Biophys. Res. Commun. 2013, 431, 496–500. [Google Scholar] [CrossRef]
- Mietus-Snyder, M.; Gowri, M.S.; Pitas, R.E. Class A Scavenger Receptor Up-Regulation in Smooth Muscle Cells by Oxidized Low Density Lipoprotein. Enhancement by Calcium Flux and Concurrent Cyclooxygenase-2 up-Regulation. J. Biol. Chem. 2000, 275, 17661–17670. [Google Scholar] [CrossRef] [Green Version]
- Zingg, J.-M.; Ricciarelli, R.; Andorno, E.; Azzi, A. Novel 5’ Exon of Scavenger Receptor CD36 Is Expressed in Cultured Human Vascular Smooth Muscle Cells and Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 412–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Yang, D.; Li, D.; Tang, B.; Yang, Y. Oleic Acid Induces Smooth Muscle Foam Cell Formation and Enhances Atherosclerotic Lesion Development via CD36. Lipids Health Dis. 2011, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Hwang, G.Y.; Liu, I.P.; Yang, V.C. Identification and Expression of Scavenger Receptor SR-BI in Endothelial Cells and Smooth Muscle Cells of Rat Aorta in Vitro and in Vivo. Atherosclerosis 2002, 161, 95–103. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kimura-Matsumoto, M.; Murakami, M.; Murakami, M.; Yamamoto, K.; Akasaka, Y.; Uzuki, M.; Yuri, Y.; Inomata, N.; Yokoo, T.; et al. Distribution of Smooth Muscle Cells and Macrophages Expressing Scavenger Receptor BI/II in Atherosclerosis. J. Atheroscler. Thromb. 2009, 16, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Chen, J.; Hermonat, P.L.; Romeo, F.; Novelli, G. Lectin-like, Oxidized Low-Density Lipoprotein Receptor-1 (LOX-1): A Critical Player in the Development of Atherosclerosis and Related Disorders. Cardiovasc. Res. 2006, 69, 36–45. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, X.; Li, N.; Dewey, W.L.; Li, P.-L.; Zhang, F. Lysosomal Cholesterol Accumulation in Macrophages Leading to Coronary Atherosclerosis in CD38(-/-) Mice. J. Cell. Mol. Med. 2016, 20, 1001–1013. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-Mediated Cholesterol Handling in Atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of Foam Cell Formation in Atherosclerosis. J. Mol. Med. Berl. Ger. 2017, 95, 1153–1165. [Google Scholar] [CrossRef]
- Hoppe, G.; O’Neil, J.; Hoff, H.F. Inactivation of Lysosomal Proteases by Oxidized Low Density Lipoprotein Is Partially Responsible for Its Poor Degradation by Mouse Peritoneal Macrophages. J. Clin. Investig. 1994, 94, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The Endoplasmic Reticulum Is the Site of Cholesterol-Induced Cytotoxicity in Macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Gonzalez, L.; Trigatti, B.L. Macrophage Apoptosis and Necrotic Core Development in Atherosclerosis: A Rapidly Advancing Field with Clinical Relevance to Imaging and Therapy. Can. J. Cardiol. 2017, 33, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.-B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noé, B. Pathology of Human Plaque Vulnerability: Mechanisms and Consequences of Intraplaque Haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic Plaque Progression and Vulnerability to Rupture: Angiogenesis as a Source of Intraplaque Hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beppu, M.; Hayashi, T.; Hasegawa, T.; Kikugawa, K. Recognition of Sialosaccharide Chains of Glycophorin on Damaged Erythrocytes by Macrophage Scavenger Receptors. Biochim. Biophys. Acta 1995, 1268, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Pasterkamp, G.; Virmani, R. The Erythrocyte: A New Player in Atheromatous Core Formation. Heart Br. Card. Soc. 2002, 88, 115–116. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hirano, K.; Nozaki, S.; Takamoto, A.; Nishida, M.; Nakagawa-Toyama, Y.; Janabi, M.Y.; Ohya, T.; Yamashita, S.; Matsuzawa, Y. Expression of Macrophage (Mphi) Scavenger Receptor, CD36, in Cultured Human Aortic Smooth Muscle Cells in Association with Expression of Peroxisome Proliferator Activated Receptor-Gamma, Which Regulates Gain of Mphi-like Phenotype in Vitro, and Its Implication in Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E Reduces the Uptake of Oxidized LDL by Inhibiting CD36 Scavenger Receptor Expression in Cultured Aortic Smooth Muscle Cells. Circulation 2000, 102, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Yuan, Z.; Wu, Y.; Liu, Y.; Zhao, Y.; Zhang, W.; Tian, Y.; Liu, W.; Liu, Y.; Kishimoto, C. High Glucose Promotes Intracellular Lipid Accumulation in Vascular Smooth Muscle Cells by Impairing Cholesterol Influx and Efflux Balance. Cardiovasc. Res. 2010, 86, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, P.J.; Greaves, D.R.; Suzuki, H.; Hakkinen, T.; Hiltunen, M.O.; Turunen, M.; Herttuala, S.Y.; Kodama, T.; Gordon, S. Analysis of Macrophage Scavenger Receptor (SR-A) Expression in Human Aortic Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.; Pitas, R.E. Synergistic Effects of Growth Factors on the Regulation of Smooth Muscle Cell Scavenger Receptor Activity. J. Biol. Chem. 1995, 270, 21672–21678. [Google Scholar] [CrossRef] [Green Version]
- Hofnagel, O.; Luechtenborg, B.; Plenz, G.; Robenek, H. Expression of the Novel Scavenger Receptor SR-PSOX in Cultured Aortic Smooth Muscle Cells and Umbilical Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 710–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wågsäter, D.; Olofsson, P.S.; Norgren, L.; Stenberg, B.; Sirsjö, A. The Chemokine and Scavenger Receptor CXCL16/SR-PSOX Is Expressed in Human Vascular Smooth Muscle Cells and Is Induced by Interferon Gamma. Biochem. Biophys. Res. Commun. 2004, 325, 1187–1193. [Google Scholar] [CrossRef]
- Luechtenborg, B.; Hofnagel, O.; Weissen-Plenz, G.; Severs, N.J.; Robenek, H. Function of Scavenger Receptor Class A Type I/II Is Not Important for Smooth Muscle Foam Cell Formation. Eur. J. Cell Biol. 2008, 87, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hofnagel, O.; Luechtenborg, B.; Stolle, K.; Lorkowski, S.; Eschert, H.; Plenz, G.; Robenek, H. Proinflammatory Cytokines Regulate LOX-1 Expression in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1789–1795. [Google Scholar] [CrossRef] [Green Version]
- Nagase, M.; Hirose, S.; Fujita, T. Unique Repetitive Sequence and Unexpected Regulation of Expression of Rat Endothelial Receptor for Oxidized Low-Density Lipoprotein (LOX-1). Biochem. J. 1998, 330 (Pt 3), 1417–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Li, D.; Sawamura, T.; Inoue, K.; Mehta, J.L. Upregulation of LOX-1 Expression in Aorta of Hypercholesterolemic Rabbits: Modulation by Losartan. Biochem. Biophys. Res. Commun. 2000, 276, 1100–1104. [Google Scholar] [CrossRef]
- Mukai, E.; Kume, N.; Hayashida, K.; Minami, M.; Yamada, Y.; Seino, Y.; Kita, T. Heparin-Binding EGF-like Growth Factor Induces Expression of Lectin-like Oxidized LDL Receptor-1 in Vascular Smooth Muscle Cells. Atherosclerosis 2004, 176, 289–296. [Google Scholar] [CrossRef]
- Bao, Z.; Li, L.; Geng, Y.; Yan, J.; Dai, Z.; Shao, C.; Sun, Z.; Jing, L.; Pang, Q.; Zhang, L.; et al. Advanced Glycation End Products Induce Vascular Smooth Muscle Cell-Derived Foam Cell Formation and Transdifferentiate to a Macrophage-Like State. Mediators Inflamm. 2020, 2020, 6850187. [Google Scholar] [CrossRef]
- Pi, S.; Mao, L.; Chen, J.; Shi, H.; Liu, Y.; Guo, X.; Li, Y.; Zhou, L.; He, H.; Yu, C.; et al. The P2RY12 Receptor Promotes VSMC-Derived Foam Cell Formation by Inhibiting Autophagy in Advanced Atherosclerosis. Autophagy 2020, 1–21. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Royo, T.; Juan-Babot, O.; Badimon, L. Adipocyte Differentiation-Related Protein Is Induced by LRP1-Mediated Aggregated LDL Internalization in Human Vascular Smooth Muscle Cells and Macrophages. J. Lipid Res. 2007, 48, 2133–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costales, P.; Fuentes-Prior, P.; Castellano, J.; Revuelta-Lopez, E.; Corral-Rodríguez, M.Á.; Nasarre, L.; Badimon, L.; Llorente-Cortes, V. K Domain CR9 of Low Density Lipoprotein (LDL) Receptor-Related Protein 1 (LRP1) Is Critical for Aggregated LDL-Induced Foam Cell Formation from Human Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 14852–14865. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, Y.; Liang, C.-P.; Thorp, E.B.; Han, S.; Jehle, A.W.; Saraswathi, V.; Pridgen, B.; Kanter, J.E.; Li, R.; et al. Defective Phagocytosis of Apoptotic Cells by Macrophages in Atherosclerotic Lesions of Ob/Ob Mice and Reversal by a Fish Oil Diet. Circ. Res. 2009, 105, 1072–1082. [Google Scholar] [CrossRef]
- Seimon, T.; Tabas, I. Mechanisms and Consequences of Macrophage Apoptosis in Atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.R.; Gibson, D.F.; Schwartz, S.M.; Tait, J.F. Binding and Phagocytosis of Apoptotic Vascular Smooth Muscle Cells Is Mediated in Part by Exposure of Phosphatidylserine. Circ. Res. 1995, 77, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.H.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular Smooth Muscle Cell Apoptosis Induces Interleukin-1-Directed Inflammation: Effects of Hyperlipidemia-Mediated Inhibition of Phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, D.M.; Lightfoot, R.; Koval, M.; Ischiropoulos, H. Autologous Apoptotic Cell Engulfment Stimulates Chemokine Secretion by Vascular Smooth Muscle Cells. Am. J. Pathol. 2005, 167, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol Loading Reprograms the MicroRNA-143/145-Myocardin Axis to Convert Aortic Smooth Muscle Cells to a Dysfunctional Macrophage-like Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.C.H.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of Vascular Smooth Muscle Cells Induces Features of Plaque Vulnerability in Atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Clarke, M.C.H.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic Apoptosis of Vascular Smooth Muscle Cells Accelerates Atherosclerosis and Promotes Calcification and Medial Degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Stoneman, V.; Braganza, D.; Figg, N.; Mercer, J.; Lang, R.; Goddard, M.; Bennett, M. Monocyte/Macrophage Suppression in CD11b Diphtheria Toxin Receptor Transgenic Mice Differentially Affects Atherogenesis and Established Plaques. Circ. Res. 2007, 100, 884–893. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nanda, V.; Direnzo, D.; Ye, J.; Xiao, S.; Kojima, Y.; Howe, K.L.; Jarr, K.-U.; Flores, A.M.; Tsantilas, P.; et al. Clonally Expanding Smooth Muscle Cells Promote Atherosclerosis by Escaping Efferocytosis and Activating the Complement Cascade. Proc. Natl. Acad. Sci. USA 2020, 117, 15818–15826. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jørgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent Intimal Foam Cells Are Deleterious at All Stages of Atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Tumurkhuu, G.; Shimada, K.; Dagvadorj, J.; Crother, T.R.; Zhang, W.; Luthringer, D.; Gottlieb, R.A.; Chen, S.; Arditi, M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res. 2016, 119, e76–e90. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular Pathology. As Based upon Physiological and Pathological Histology. Lecture XVI—Atheromatous Affection of Arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional Accumulations of T Cells, Macrophages, and Smooth Muscle Cells in the Human Atherosclerotic Plaque. Arterioscler. Dallas Tex 1986, 6, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Hoff, H.F.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Stimulation of Cholesteryl Ester Synthesis in Macrophages by Extracts of Atherosclerotic Human Aortas and Complexes of Albumin/Cholesteryl Esters. Arterioscler. Dallas Tex 1981, 1, 210–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.-A.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-Cell Immune Landscape of Human Atherosclerotic Plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.; de Winther, M.P. Macrophage Subsets in Atherosclerosis as Defined by Single-Cell Technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Classics in Arteriosclerosis Research: On Experimental Cholesterin Steatosis and Its Significance in the Origin of Some Pathological Processes by N. Anitschkow and S. Chalatow, Translated by Mary Z. Pelias, 1913. Arterioscler. Dallas Tex. 1983, 3, 178–182.
- Herijgers, N.; Van Eck, M.; Groot, P.H.E.; Hoogerbrugge, P.M.; Van Berkel, T.J.C. Effect of Bone Marrow Transplantation on Lipoprotein Metabolism and Atherosclerosis in LDL Receptor—Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1995–2003. [Google Scholar] [CrossRef]
- Kennedy, A.; Gruen, M.L.; Gutierrez, D.A.; Surmi, B.K.; Orr, J.S.; Webb, C.D.; Hasty, A.H. Impact of Macrophage Inflammatory Protein-1α Deficiency on Atherosclerotic Lesion Formation, Hepatic Steatosis, and Adipose Tissue Expansion. PLoS ONE 2012, 7, e31508. [Google Scholar] [CrossRef]
- Kubo, N.; McCurdy, S.; Boisvert, W.A. Defective Fas Expression on Bone Marrow Derived Cells Alters Atherosclerotic Plaque Morphology in Hyperlipidemic Mice. Discov. Craiova Rom. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Nong, Z.; Gonzalez-Navarro, H.; Amar, M.; Freeman, L.; Knapper, C.; Neufeld, E.B.; Paigen, B.J.; Hoyt, R.F.; Fruchart-Najib, J.; Santamarina-Fojo, S. Hepatic Lipase Expression in Macrophages Contributes to Atherosclerosis in ApoE-Deficient and LCAT-Transgenic Mice. J. Clin. Investig. 2003, 112, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Fazio, S.; Linton, M.F. Interplay between Apolipoprotein E and Scavenger Receptor Class B Type I Controls Coronary Atherosclerosis and Lifespan in the Mouse. Circulation 2005, 111, 3349–3351. [Google Scholar] [CrossRef] [Green Version]
- Lessner, S.M.; Prado, H.L.; Waller, E.K.; Galis, Z.S. Atherosclerotic Lesions Grow through Recruitment and Proliferation of Circulating Monocytes in a Murine Model. Am. J. Pathol. 2002, 160, 2145–2155. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.-E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Pease, D.C.; Paule, W.J. Electron Microscopy of Elastic Arteries; the Thoracic Aorta of the Rat. J. Ultrastruct. Res. 1960, 3, 469–483. [Google Scholar] [CrossRef]
- Parker, F. An Electron Microscopic Study of Experimental Atherosclerosis. Am. J. Pathol. 1960, 36, 19–53. [Google Scholar] [PubMed]
- Imai, H.; Lee, K.T.; Pastori, S.; Panlilio, E.; Florentin, R.; Thomas, W.A. Atherosclerosis in Rabbits. Architectural and Subcellular Alterations of Smooth Muscle Cells of Aortas in Response to Hyperlipemia. Exp. Mol. Pathol. 1966, 5, 273–310. [Google Scholar] [CrossRef]
- Wissler, R.W. The Arterial Medial Cell, Smooth Muscle, or Multifunctional Mesenchyme? Circulation 1967, 36, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Aqel, N.M.; Ball, R.Y.; Waldmann, H.; Mitchinson, M.J. Monocytic Origin of Foam Cells in Human Atherosclerotic Plaques. Atherosclerosis 1984, 53, 265–271. [Google Scholar] [CrossRef]
- Aqel, N.M.; Ball, R.Y.; Waldmann, H.; Mitchinson, M.J. Identification of Macrophages and Smooth Muscle Cells in Human Atherosclerosis Using Monoclonal Antibodies. J. Pathol. 1985, 146, 197–204. [Google Scholar] [CrossRef]
- Watanabe, T.; Hirata, M.; Yoshikawa, Y.; Nagafuchi, Y.; Toyoshima, H.; Watanabe, T. Role of Macrophages in Atherosclerosis. Sequential Observations of Cholesterol-Induced Rabbit Aortic Lesion by the Immunoperoxidase Technique Using Monoclonal Antimacrophage Antibody. Lab. Investig. J. Tech. Methods Pathol. 1985, 53, 80–90. [Google Scholar]
- Roessner, A.; Herrera, A.; Höning, H.J.; Vollmer, E.; Zwadlo, G.; Schürmann, R.; Sorg, C.; Grundmann, E. Identification of Macrophages and Smooth Muscle Cells with Monoclonal Antibodies in the Human Atherosclerotic Plaque. Virchows Arch. A Pathol. Anat. Histopathol. 1987, 412, 169–174. [Google Scholar] [CrossRef]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The Pathogenesis of Atherosclerosis: A Perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Gordon, D.; San, H.; Pompili, V.J.; Imperiale, M.J.; Nabel, G.J.; Nabel, E.G. Gene Therapy for Vascular Smooth Muscle Cell Proliferation after Arterial Injury. Science 1994, 265, 781–784. [Google Scholar] [CrossRef]
- Stemerman, M.B.; Ross, R. Experimental Arteriosclerosis. I. Fibrous Plaque Formation in Primates, an Electron Microscope Study. J. Exp. Med. 1972, 136, 769–789. [Google Scholar] [CrossRef] [Green Version]
- Bart, R.D.; Sheng, H.; Laskowitz, D.T.; Pearlstein, R.D.; Warner, D.S. Regional CBF in Apolipoprotein E-Deficient and Wild Type Mice during Focal Cerebral Ischemia. Neuroreport 1998, 9, 2615–2620. [Google Scholar] [CrossRef]
- Lupieri, A.; Smirnova, N.F.; Solinhac, R.; Malet, N.; Benamar, M.; Saoudi, A.; Santos-Zas, I.; Zeboudj, L.; Ait-Oufella, H.; Hirsch, E.; et al. Smooth Muscle Cells-Derived CXCL10 Prevents Endothelial Healing through PI3Kγ-Dependent T Cells Response. Cardiovasc. Res. 2020, 116, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma Lipid Profiling across Species for the Identification of Optimal Animal Models of Human Dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.S.; Reardon, C.A. Diet and Murine Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal Models of Atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Lally, J.I. The Activity of an Esterified Cholesterol Transferring Factor in Human and Rat Serum. Biochim. Biophys. Acta 1978, 531, 233–236. [Google Scholar] [CrossRef]
- Daugherty, A. Mouse Models of Atherosclerosis. Am. J. Med. Sci. 2002, 323, 3–10. [Google Scholar] [CrossRef]
- Von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017, 25, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Bartoli-Leonard, F.; Aikawa, E. Recapitulating the Complex Pathology of Atherosclerosis: Which Model to Use? Circ. Res. 2021, 129, 491–493. [Google Scholar] [CrossRef]
- Katsuda, S.; Okada, Y.; Minamoto, T.; Oda, Y.; Matsui, Y.; Nakanishi, I. Collagens in Human Atherosclerosis. Immunohistochemical Analysis Using Collagen Type-Specific Antibodies. Arterioscler. Thromb. J. Vasc. Biol. 1992, 12, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Rekhter, M.D.; Zhang, K.; Narayanan, A.S.; Phan, S.; Schork, M.A.; Gordon, D. Type I Collagen Gene Expression in Human Atherosclerosis. Localization to Specific Plaque Regions. Am. J. Pathol. 1993, 143, 1634–1648. [Google Scholar]
- Korol, R.M.; Canham, P.B.; Liu, L.; Viswanathan, K.; Ferguson, G.G.; Hammond, R.R.; Finlay, H.M.; Baker, H.V.; Lopez, C.; Lucas, A.R. Detection of Altered Extracellular Matrix in Surface Layers of Unstable Carotid Plaque: An Optical Spectroscopy, Birefringence and Microarray Genetic Analysis. Photochem. Photobiol. 2011, 87, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Sukhova, G.; Lee, R.T.; Liao, J.K. Molecular Biology of Atherosclerosis. Int. J. Cardiol. 1997, 62 (Suppl. 2), S23–S29. [Google Scholar] [CrossRef]
- Libby, P.; Schoenbeck, U.; Mach, F.; Selwyn, A.P.; Ganz, P. Current Concepts in Cardiovascular Pathology: The Role of LDL Cholesterol in Plaque Rupture and Stabilization. Am. J. Med. 1998, 104, 14S–18S. [Google Scholar] [CrossRef]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased Expression of Matrix Metalloproteinases and Matrix Degrading Activity in Vulnerable Regions of Human Atherosclerotic Plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galis, Z.S.; Sukhova, G.K.; Kranzhöfer, R.; Clark, S.; Libby, P. Macrophage Foam Cells from Experimental Atheroma Constitutively Produce Matrix-Degrading Proteinases. Proc. Natl. Acad. Sci. USA 1995, 92, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.J.; Bond, M.; Crook, M.F.; Newby, A.C. Role of Nuclear Factor-Kappa B Activation in Metalloproteinase-1, -3, and -9 Secretion by Human Macrophages in Vitro and Rabbit Foam Cells Produced in Vivo. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-B.; Yang, B.; Li, X.; Liu, H.; Lei, G. Lysophosphatidic Acid Promotes Expression and Activation of Matrix Metalloproteinase 9 (MMP9) in THP-1 Cells via Toll-Like Receptor 4/Nuclear Factor-ΚB (TLR4/NF-ΚB) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 4861–4868. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinase Production from Macrophages—APerfect Storm Leading to Atherosclerotic Plaque Rupture and Myocardial Infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Waqar, A.B.; Nishijima, K.; Ning, B.; Kitajima, S.; Matsuhisa, F.; Chen, L.; Liu, E.; Koike, T.; Yu, Y.; et al. Macrophage-Derived MMP-9 Enhances the Progression of Atherosclerotic Lesions and Vascular Calcification in Transgenic Rabbits. J. Cell. Mol. Med. 2020, 24, 4261–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Zhang, F.; Grassia, G.; Hu, Y.; Zhang, Z.; Xing, Q.; Yin, X.; Maddaluno, M.; Drung, B.; Schmidt, B.; et al. Matrix Metalloproteinase-8 Promotes Vascular Smooth Muscle Cell Proliferation and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Chen, Q.; Yang, M.; Maguire, E.M.; Yu, X.; He, S.; Xiao, R.; Wang, C.S.; An, W.; Wu, W.; et al. Macrophage-Derived MMP-8 Determines Smooth Muscle Cell Differentiation from Adventitia Stem/Progenitor Cells and Promotes Neointima Hyperplasia. Cardiovasc. Res. 2020, 116, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Guadagni, F. Protease Nexin-1: A Regulator of Vascular Disease? J. Thromb. Haemost. JTH 2006, 4, 320–321. [Google Scholar] [CrossRef]
- Mansilla, S.; Boulaftali, Y.; Venisse, L.; Arocas, V.; Meilhac, O.; Michel, J.-B.; Jandrot-Perrus, M.; Bouton, M.-C. Macrophages and Platelets Are the Major Source of Protease Nexin-1 in Human Atherosclerotic Plaque. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1844–1850. [Google Scholar] [CrossRef]
- Cho, K.Y.; Miyoshi, H.; Kuroda, S.; Yasuda, H.; Kamiyama, K.; Nakagawara, J.; Takigami, M.; Kondo, T.; Atsumi, T. The Phenotype of Infiltrating Macrophages Influences Arteriosclerotic Plaque Vulnerability in the Carotid Artery. J. Stroke Cerebrovasc. Dis. 2013, 22, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Farb, A.; Malcom, G.T.; Liang, Y.H.; Smialek, J.; Virmani, R. Coronary Risk Factors and Plaque Morphology in Men with Coronary Disease Who Died Suddenly. N. Engl. J. Med. 1997, 336, 1276–1282. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the Vulnerable Plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.; Brittenden, J.; Lahiri, R.; Brown, P.A.J.; Thies, F.; Wilson, H.M. Macrophage Subtypes in Symptomatic Carotid Artery and Femoral Artery Plaques. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2012, 44, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Cosentino, N.; Fiorelli, S.; Fabbiocchi, F.; Niccoli, G.; Refaat, H.; Camera, M.; Calligaris, G.; De Martini, S.; Bonomi, A.; et al. Biological Profile of Monocyte-Derived Macrophages in Coronary Heart Disease Patients: Implications for Plaque Morphology. Sci. Rep. 2019, 9, 8680. [Google Scholar] [CrossRef]
- Davies, M.J.; Richardson, P.D.; Woolf, N.; Katz, D.R.; Mann, J. Risk of Thrombosis in Human Atherosclerotic Plaques: Role of Extracellular Lipid, Macrophage, and Smooth Muscle Cell Content. Br. Heart J. 1993, 69, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Gomez, D.; Shankman, L.S.; Swiatlowska, P.; Williams, J.; Sarmento, O.F.; Alencar, G.F.; Hess, D.L.; Bevard, M.H.; Greene, E.S.; et al. Activation of the Pluripotency Factor OCT4 in Smooth Muscle Cells Is Atheroprotective. Nat. Med. 2016, 22, 657–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β Has Atheroprotective Effects in Advanced Atherosclerotic Lesions of Mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the Atherosclerotic Plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Peeters, W.; Busser, E.; Lamers, D.; Sluijter, J.P.G.; Goumans, M.-J.; de Weger, R.A.; Moll, F.L.; Doevendans, P.A.; Pasterkamp, G.; et al. Balance between Angiopoietin-1 and Angiopoietin-2 Is in Favor of Angiopoietin-2 in Atherosclerotic Plaques with High Microvessel Density. J. Vasc. Res. 2008, 45, 244–250. [Google Scholar] [CrossRef]
- Michel, J.-B.; Virmani, R.; Arbustini, E.; Pasterkamp, G. Intraplaque Haemorrhages as the Trigger of Plaque Vulnerability. Eur. Heart J. 2011, 32, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin Directs Macrophage Differentiation and Prevents Foam Cell Formation in Human Atherosclerotic Plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higginbotham, A.C.; Higginbotham, F.H. The Relation of Plaque Composition and of the Vasa Vasorum to Vascularization of Atheromatous Plaques in Rabbits and Man. J. Atheroscler. Res. 1961, 1, 283–295. [Google Scholar] [CrossRef]
- Pelisek, J.; Well, G.; Reeps, C.; Rudelius, M.; Kuehnl, A.; Culmes, M.; Poppert, H.; Zimmermann, A.; Berger, H.; Eckstein, H.-H. Neovascularization and Angiogenic Factors in Advanced Human Carotid Artery Stenosis. Circ. J. Off. J. Jpn. Circ. Soc. 2012, 76, 1274–1282. [Google Scholar] [CrossRef] [Green Version]
- Ho-Tin-Noé, B.; Le Dall, J.; Gomez, D.; Louedec, L.; Vranckx, R.; El-Bouchtaoui, M.; Legrès, L.; Meilhac, O.; Michel, J.-B. Early Atheroma-Derived Agonists of Peroxisome Proliferator-Activated Receptor-γ Trigger Intramedial Angiogenesis in a Smooth Muscle Cell-Dependent Manner. Circ. Res. 2011, 109, 1003–1014. [Google Scholar] [CrossRef]
- Ferrara, N.; Winer, J.; Burton, T. Aortic Smooth Muscle Cells Express and Secrete Vascular Endothelial Growth Factor. Growth Factors Chur Switz. 1991, 5, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, M.; Satake, S.; Esaki, T.; Yamada, K.; Hayashi, T.; Naito, M.; Asai, K.; Iguchi, A. Induction of Angiogenesis by Smooth Muscle Cell-Derived Factor: Possible Role in Neovascularization in Atherosclerotic Plaque. J. Cell. Physiol. 1995, 164, 658–667. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/Macrophage Regulation of Vascular Calcification in Vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef]
- Champagne, C.M.; Takebe, J.; Offenbacher, S.; Cooper, L.F. Macrophage Cell Lines Produce Osteoinductive Signals That Include Bone Morphogenetic Protein-2. Bone 2002, 30, 26–31. [Google Scholar] [CrossRef]
- Ikeda, K.; Souma, Y.; Akakabe, Y.; Kitamura, Y.; Matsuo, K.; Shimoda, Y.; Ueyama, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Macrophages Play a Unique Role in the Plaque Calcification by Enhancing the Osteogenic Signals Exerted by Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2012, 425, 39–44. [Google Scholar] [CrossRef]
- Dai, J.; Tian, J.; Hou, J.; Xing, L.; Liu, S.; Ma, L.; Yu, H.; Ren, X.; Dong, N.; Yu, B. Association between Cholesterol Crystals and Culprit Lesion Vulnerability in Patients with Acute Coronary Syndrome: An Optical Coherence Tomography Study. Atherosclerosis 2016, 247, 111–117. [Google Scholar] [CrossRef]
- Shi, X.; Cai, H.; Wang, F.; Liu, R.; Xu, X.; Li, M.; Han, Y.; Yin, Q.; Ye, R.; Liu, X. Cholesterol Crystals Are Associated with Carotid Plaque Vulnerability: An Optical Coherence Tomography Study. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2020, 29, 104579. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Dai, J.; Zhang, S.; Wang, Y.; Wang, J.; Li, L.; Yu, H.; Wei, G.; Zhang, X.; Feng, N.; et al. Culprit Lesion Morphology in Young Patients with ST-Segment Elevated Myocardial Infarction: A Clinical, Angiographic and Optical Coherence Tomography Study. Atherosclerosis 2019, 289, 94–100. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic Control of Smooth Muscle Cell Differentiation and Phenotypic Switching in Vascular Development and Disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Swiatlowska, P.; Owens, G.K. Epigenetic Control of Smooth Muscle Cell Identity and Lineage Memory. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2508–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebboudj, A.F.; Imura, M.; Boström, K. Matrix GLA Protein, a Regulatory Protein for Bone Morphogenetic Protein-2. J. Biol. Chem. 2002, 277, 4388–4394. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the Gene Encoding the Human Matrix Gla Protein Cause Keutel Syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The Antioxidant Tempol Ameliorates Arterial Medial Calcification in Uremic Rats: Important Role of Oxidative Stress in the Pathogenesis of Vascular Calcification in Chronic Kidney Disease. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef]
- Nakano-Kurimoto, R.; Ikeda, K.; Uraoka, M.; Nakagawa, Y.; Yutaka, K.; Koide, M.; Takahashi, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Replicative Senescence of Vascular Smooth Muscle Cells Enhances the Calcification through Initiating the Osteoblastic Transition. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1673–H1684. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.C.; Nesmith, A.P.; Parker, K.K. The Role of Mechanotransduction on Vascular Smooth Muscle Myocytes’ [Corrected] Cytoskeleton and Contractile Function. Anat. Rec. Hoboken NJ 2007 2014, 297, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Pepper, J. Differential Aspects of the Disease and Treatment of Thoracic Acute Aortic Dissection (TAAD)-the European Experience. Ann. Cardiothorac. Surg. 2016, 5, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. JASN 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis Accelerates Medial Vascular Calcification in Part by Triggering Smooth Muscle Cell Apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Remaley, A.T.; Sviridov, D. HDL Therapy: Two Kinds of Right? Curr. Pharm. Des. 2010, 16, 4134–4147. [Google Scholar] [CrossRef]
- Yamashita, S.; Ruscica, M.; Macchi, C.; Corsini, A.; Matsuzawa, Y.; Sirtori, C.R. Cholesteryl Ester Transfer Protein: An Enigmatic Pharmacology—Antagonists and Agonists. Atherosclerosis 2018, 278, 286–298. [Google Scholar] [CrossRef]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.S.; Didonato, J.A.; et al. Effects of Native and Myeloperoxidase-Modified Apolipoprotein a-I on Reverse Cholesterol Transport and Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef]
- Sanson, M.; Distel, E.; Fisher, E.A. HDL Induces the Expression of the M2 Macrophage Markers Arginase 1 and Fizz-1 in a STAT6-Dependent Process. PLoS ONE 2013, 8, e74676. [Google Scholar] [CrossRef]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL Promotes Rapid Atherosclerosis Regression in Mice and Alters Inflammatory Properties of Plaque Monocyte-Derived Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.J.; Distel, E.; Murphy, A.J.; Hu, J.; Garshick, M.S.; Ogando, Y.; Liu, J.; Vaisar, T.; Heinecke, J.W.; Berger, J.S.; et al. Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation 2019, 140, 1170–1184. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Andrews, J.; Kastelein, J.J.P.; Merkely, B.; Nissen, S.E.; Ray, K.K.; Schwartz, G.G.; Worthley, S.G.; Keyserling, C.; Dasseux, J.-L.; et al. Effect of Serial Infusions of CER-001, a Pre-β High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.H.; Kaiser, Y.; van Olden, C.C.; Santos, R.D.; Dasseux, J.-L.; Genest, J.; Gaudet, D.; Westerink, J.; Keyserling, C.; Verberne, H.J.; et al. No Benefit of HDL Mimetic CER-001 on Carotid Atherosclerosis in Patients with Genetically Determined Very Low HDL Levels. Atherosclerosis 2020, 311, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Amengual, J.; Menon, A.; Kamaly, N.; Zhou, F.; Xu, X.; Saw, P.E.; Lee, S.-J.; Si, K.; Ortega, C.A.; et al. Targeted Nanotherapeutics Encapsulating Liver X Receptor Agonist GW3965 Enhance Antiatherogenic Effects without Adverse Effects on Hepatic Lipid Metabolism in Ldlr-/- Mice. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, W.; Yu, B.; Kuai, R.; Hu, W.; Morin, E.E.; Garcia-Barrio, M.T.; Zhang, J.; Moon, J.J.; Schwendeman, A.; et al. Synthetic High-Density Lipoprotein-Mediated Targeted Delivery of Liver X Receptors Agonist Promotes Atherosclerosis Regression. EBioMedicine 2018, 28, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.Y.; Rahmani, M.; Wong, B.W.; Allahverdian, S.; McManus, B.M.; Pickering, J.G.; Chan, T.; Francis, G.A. ATP-Binding Cassette Transporter A1 Expression and Apolipoprotein A-I Binding Are Impaired in Intima-Type Arterial Smooth Muscle Cells. Circulation 2009, 119, 3223–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzych, J.M.; Pearce, E.; Cheever, A.; Caulada, Z.A.; Caspar, P.; Heiny, S.; Lewis, F.; Sher, A. Egg Deposition Is the Major Stimulus for the Production of Th2 Cytokines in Murine Schistosomiasis Mansoni. J. Immunol. Baltim. Md 1950 1991, 146, 1322–1327. [Google Scholar]
- Wolfs, I.M.J.; Stöger, J.L.; Goossens, P.; Pöttgens, C.; Gijbels, M.J.J.; Wijnands, E.; van der Vorst, E.P.C.; van Gorp, P.; Beckers, L.; Engel, D.; et al. Reprogramming Macrophages to an Anti-Inflammatory Phenotype by Helminth Antigens Reduces Murine Atherosclerosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jørgensen, H.F. Disease-Relevant Transcriptional Signatures Identified in Individual Smooth Muscle Cells from Healthy Mouse Vessels. Nat. Commun. 2018, 9, 4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective Roles of Smooth Muscle Cell Phenotypic Modulation and the TCF21 Disease Gene as Revealed by Single-Cell Analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Hartman, R.J.G.; Owsiany, K.; Ma, L.; Koplev, S.; Hao, K.; Slenders, L.; Civelek, M.; Mokry, M.; Kovacic, J.C.; Pasterkamp, G.; et al. Sex-Stratified Gene Regulatory Networks Reveal Female Key Driver Genes of Atherosclerosis Involved in Smooth Muscle Cell Phenotype Switching. Circulation 2021, 143, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [Green Version]

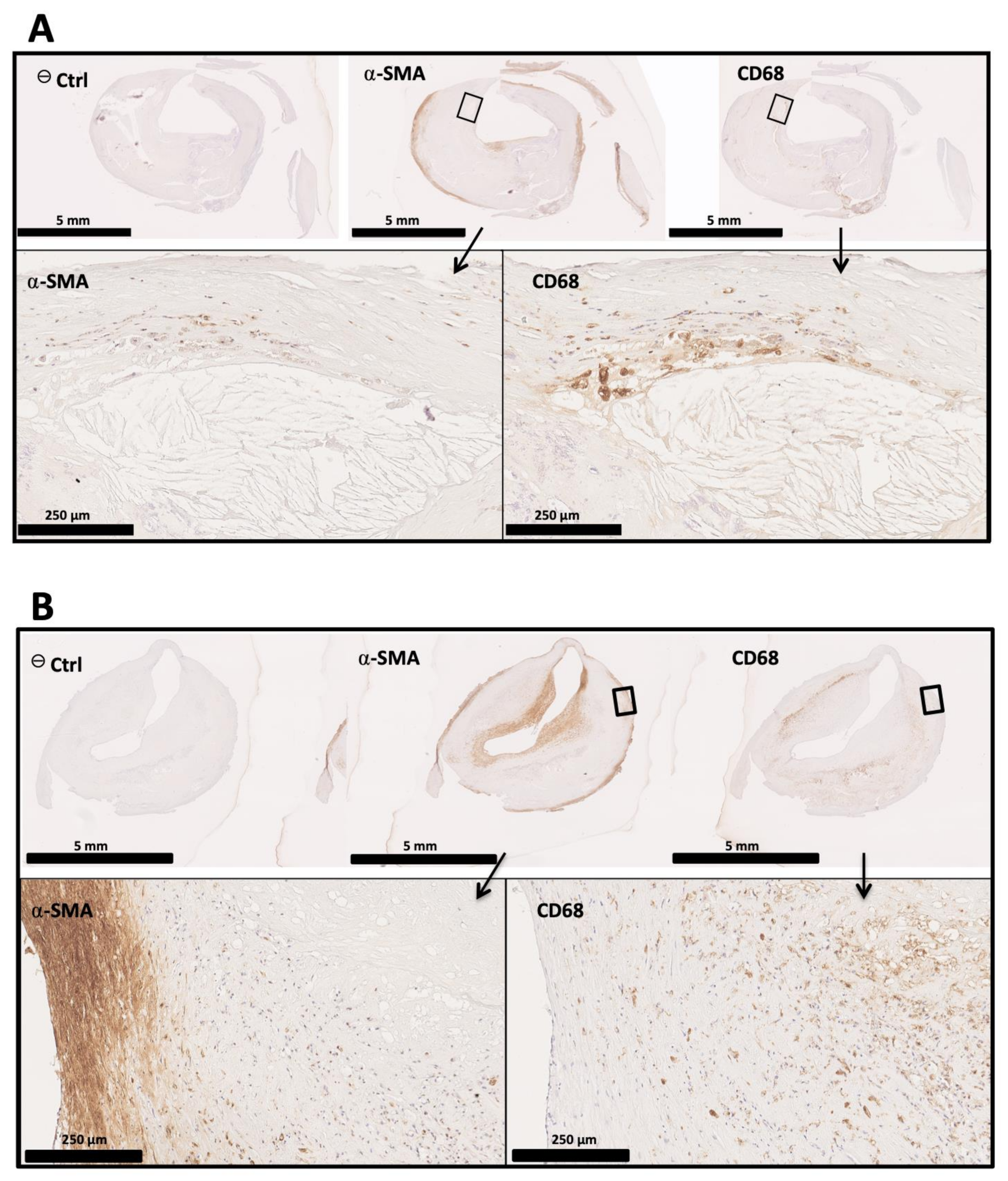
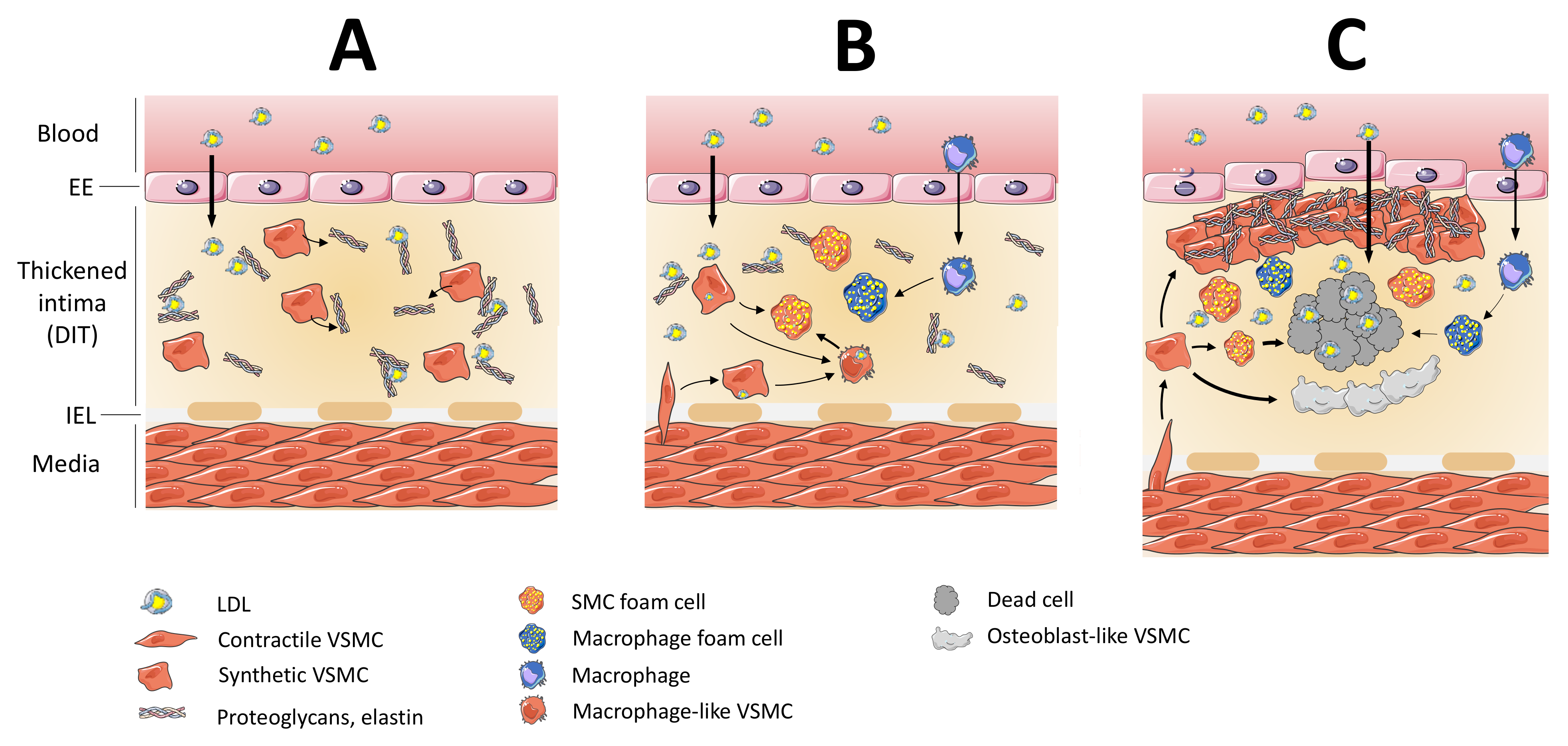

{kind=link}
{kind=link}
{kind=link}
| Cell Type | Receptor | Stimulus | Function | References |
|---|---|---|---|---|
| Macrophages | Scavenger receptor-A1 (SR-A1) | Visfatin, ox-LDL | Ox-LDL uptake, cell apoptosis, foam cell formation | [127,128,129] |
| Lectin-like oxidized LDL receptor-1 (LOX-1) | Ox-LDL, ER stress, modified lipoproteins, advanced glycation end-products (AGEs) | Ox-LDL uptake, inhibition of macrophage migration, increase in foam cell formation | [130,131,132,133] | |
| CD 36 | Ox-LDL, ox-phospholipids, IL-34, visfatin | Ox-LDL uptake, cytokine release, NLRP3 inflammasome activation, increased in foam cell formation | [130,131,134,135] | |
| CD 68 | Ox-LDL | Ox-LDL uptake, contribution to inflammation | [136,137] | |
| CD 146 | Ox-LDL | Foam cell formation, macrophage retention | [138] | |
| TLR4 | Ox-LDL, LPS, saturated fatty acids | Mediates inflammatory response | [134,135,139] | |
| Macrophage scavenger receptor -1 (MSR-1) | Native or modified LDL (acetylated, oxidized | Ox-LDL uptake | [140,141] | |
| SMC | Scavenger receptor-A1 (SR-A1) | Phorbol esters, ROS, ox-LDL | Ox-LDL uptake, foam cell formation | [142,143] |
| CD 36 | Free fatty acids (Oleic acid), ox-LDL | Free fatty acid and ox-LDL uptake, Foam cell formation | [144] | |
| Scavenger Receptor BI/II | Intracellular cholesterol content | Cholesterol ester uptake, apoB-containing lipoproteins | [145,146] | |
| Lectin-like oxidized LDL receptor-1 (LOX-1) | LPS, TNF-a, IL-1β, IFN-y, ox-LDL, shear stress | SMC proliferation, ox-LDL uptake, foam cell formation | [131,147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Checkouri, E.; Blanchard, V.; Meilhac, O. Macrophages in Atherosclerosis, First or Second Row Players? Biomedicines 2021, 9, 1214. https://doi.org/10.3390/biomedicines9091214
Checkouri E, Blanchard V, Meilhac O. Macrophages in Atherosclerosis, First or Second Row Players? Biomedicines. 2021; 9(9):1214. https://doi.org/10.3390/biomedicines9091214
Chicago/Turabian StyleCheckouri, Eloïse, Valentin Blanchard, and Olivier Meilhac. 2021. "Macrophages in Atherosclerosis, First or Second Row Players?" Biomedicines 9, no. 9: 1214. https://doi.org/10.3390/biomedicines9091214
APA StyleCheckouri, E., Blanchard, V., & Meilhac, O. (2021). Macrophages in Atherosclerosis, First or Second Row Players? Biomedicines, 9(9), 1214. https://doi.org/10.3390/biomedicines9091214