
Inhibition of Aminoglycoside 6′-N-acetyltransferase Type Ib (AAC(6′)-Ib): Structure–Activity Relationship of Substituted Pyrrolidine Pentamine Derivatives as Inhibitors

 , , ,
, , , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Cultures
2.2. Synthesis of Small Molecule Compounds
2.3. High-Performance Liquid Chromatography (HPLC) Purification
2.4. Liquid Chromatography–Mass Spectrometry (LCMS) Analysis of Purified Material
2.5. Initial Growth Inhibition Assays
2.6. Modeling
2.7. Checkerboard Assays
3. Results
3.1. Synthesis and Preliminary Analysis of Analogs to Compound 2637.001
3.2. Molecular Docking
3.3. Potentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Man, T.J.B.; Lutgring, J.D.; Lonsway, D.R.; Anderson, K.F.; Kiehlbauch, J.A.; Chen, L.; Walters, M.S.; Sjolund-Karlsson, M.; Rasheed, J.K.; Kallen, A.; et al. Genomic analysis of a pan-resistant isolate of Klebsiella pneumoniae, United States 2016. mBio 2018, 9, e00440-18. [Google Scholar] [CrossRef] [Green Version]
- Boucher, H.W. Bad bugs, no drugs 2002–2020: Progress, challenges, and call to action. Trans. Am. Clin. Climatol Assoc. 2020, 131, 65–71. [Google Scholar]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017; Available online: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/ (accessed on 1 September 2021).
- Tolmasky, M.E. Strategies to prolong the useful life of existing antibiotics and help overcoming the antibiotic resistance crisis. In Frontiers in Clinical Drug Research-Anti Infectives; Rhaman, A., Ed.; Bentham Books: Sharjah, United Arab Emirates, 2017; Volume 1, pp. 1–27. [Google Scholar]
- Papp-Wallace, K.M.; Bonomo, R.A. New beta-lactamase inhibitors in the clinic. Infect. Dis. Clin. N. Am. 2016, 30, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.S.; Bonomo, R.A.; Tolmasky, M.E. Carbapenemases: Transforming Acinetobacter baumannii into a yet more dangerous menace. Biomolecules 2020, 10, 720. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Moellering, R. Antibacterial agents. In Manual of Clinical Microbiology; Murray, P., Baron, E., Jorgensen, J., Landry, M., Pfaller, M., Eds.; American Society for Microbiology Press: Washington, DC, USA, 2007; Volume 1, pp. 1077–1113. [Google Scholar]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. Medchemcomm 2016, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and dissemination of aminoglycoside resistance: The aac(6′)-Ib paradigm. Front. Microbiol. 2013, 4, 121. [Google Scholar] [CrossRef] [Green Version]
- Yarlagadda, V.; Medina, R.; Wright, G.D. Venturicidin A, A membrane-active natural product inhibitor of ATP synthase potentiates aminoglycoside antibiotics. Sci. Rep. 2020, 10, 8134. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Labby, K.J.; Garneau-Tsodikova, S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013, 5, 1285–1309. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Caldwell, S.J.; Fong, D.H.; Berghuis, A.M. Prospects for circumventing aminoglycoside kinase mediated antibiotic resistance. Front. Cell Infect. Microbiol. 2013, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Vong, K.; Wee, K.; Fakhoury, J.; Dullaghan, E.; Auclair, K. Cellular studies of an aminoglycoside potentiator reveal a new inhibitor of aminoglycoside resistance. ChemBioChem 2018, 19, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, L.; De Oliveira, D.M.P.; El-Deeb, I.M.; Brazel, E.B.; Harbison-Price, N.; Ong, C.Y.; Rivera-Hernandez, T.; Ferguson, S.A.; Cork, A.J.; Phan, M.D.; et al. Chemical synergy between ionophore PBT2 and zinc reverses antibiotic resistance. mBio 2018, 9, e02391-18. [Google Scholar] [CrossRef] [Green Version]
- Chiem, K.; Fuentes, B.A.; Lin, D.L.; Tran, T.; Jackson, A.; Ramirez, M.S.; Tolmasky, M.E. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib-mediated amikacin resistance in Klebsiella pneumoniae by zinc and copper pyrithione. Antimicrob. Agents Chemother. 2015, 59, 5851–5853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiem, K.; Hue, F.; Magallon, J.; Tolmasky, M.E. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib-mediated amikacin resistance by zinc complexed with clioquinol, an ionophore active against tumors and neurodegenerative diseases. Int. J. Antimicrob. Agents 2018, 51, 271–273. [Google Scholar] [CrossRef]
- Lin, D.L.; Tran, T.; Alam, J.Y.; Herron, S.R.; Ramirez, M.S.; Tolmasky, M.E. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib by zinc: Reversal of amikacin resistance in Acinetobacter baumannii and Escherichia coli by a zinc ionophore. Antimicrob. Agents Chemother. 2014, 58, 4238–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiem, K.; Jani, S.; Fuentes, B.; Lin, D.L.; Rasche, M.E.; Tolmasky, M.E. Identification of an inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] by glide molecular docking. Medchemcomm 2016, 7, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.L.; Tran, T.; Adams, C.; Alam, J.Y.; Herron, S.R.; Tolmasky, M.E. Inhibitors of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] identified by in silico molecular docking. Bioorg. Med. Chem. Lett. 2013, 23, 5694–5698. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.; Chiem, K.; Jani, S.; Arivett, B.A.; Lin, D.L.; Lad, R.; Jimenez, V.; Farone, M.B.; Debevec, G.; Santos, R.; et al. Identification of a small molecule inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] using mixture-based combinatorial libraries. Int. J. Antimicrob. Agents 2018, 51, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Vong, K.; Auclair, K. Understanding and overcoming aminoglycoside resistance caused by N-6′-acetyltransferase. Medchemcomm 2012, 3, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Sewunet, T.; Asrat, D.; Woldeamanuel, Y.; Ny, S.; Westerlund, F.; Aseffa, A.; Giske, C.G. High prevalence of blaCTX-M-15 and nosocomial transmission of hypervirulent epidemic clones of Klebsiella pneumoniae at a tertiary hospital in Ethiopia. JAC Antimicrob. Resist. 2021, 3, dlab001. [Google Scholar] [CrossRef]
- Vetting, M.W.; Park, C.H.; Hegde, S.S.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 2008, 47, 9825–9835. [Google Scholar] [CrossRef] [Green Version]
- Blondelle, S.E.; Pinilla, C.; Boggiano, C. Synthetic combinatorial libraries as an alternative strategy for the development of novel treatments for infectious diseases. Methods Enzymol. 2003, 369, 322–344. [Google Scholar] [PubMed]
- Ramirez, M.S.; Vilacoba, E.; Stietz, M.S.; Merkier, A.K.; Jeric, P.; Limansky, A.S.; Marquez, C.; Bello, H.; Catalano, M.; Centron, D. Spreading of AbaR-type genomic islands in multidrug resistance Acinetobacter baumannii strains belonging to different clonal complexes. Curr. Microbiol. 2013, 67, 9–14. [Google Scholar] [CrossRef]
- Lopez, C.; Arivett, B.A.; Actis, L.A.; Tolmasky, M.E. Inhibition of AAC(6′)-Ib-mediated resistance to amikacin in Acinetobacter baumannii by an antisense peptide-conjugated 2′,4′-bridged nucleic acid-NC-DNA hybrid oligomer. Antimicrob. Agents Chemother. 2015, 59, 5798–5803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arivett, B.A.; Fiester, S.E.; Ream, D.C.; Centron, D.; Ramirez, M.S.; Tolmasky, M.E.; Actis, L.A. Draft Genome of the Multidrug-Resistant Acinetobacter baumannii Strain A155 Clinical Isolate. Genome Announc. 2015, 3, e00047-20. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Hoel, D.G. Statistical aspects of chemical mixtures. In Methods for Assessing the Effects of Mixtures of Chemicals; Vouk, V.B., Butler, G.C., Upton, A.C., Parke, D.V., Asher, S.C., Eds.; Wiley: New York, NY, USA, 1987; pp. 369–377. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar]
- Boucher, H.W.; Ambrose, P.G.; Chambers, H.F.; Ebright, R.H.; Jezek, A.; Murray, B.E.; Newland, J.G.; Ostrowsky, B.; Rex, J.H.; Infectious Diseases Society of America. White paper: Developing antimicrobial drugs for resistant pathogens, narrow-spectrum indications, and unmet needs. J. Infect. Dis. 2017, 216, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.D.; Punetha, A.; Hou, C.; Garneau-Tsodikova, S.; Tsodikov, O.V. Probing the robustness of inhibitors of tuberculosis aminoglycoside resistance enzyme Eis by mutagenesis. ACS Infect. Dis. 2019, 5, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Amikacin: Uses, resistance, and prospects for inhibition. Molecules 2017, 22, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Antibiotic Resistance Threats in the United States; Centers for Disease Control: Atlanta, GA, USA, 2019.
- Magallon, J.; Vu, P.; Reeves, C.; Kwan, S.; Phan, K.; Oakley-Havens, C.; Ramirez, M.S.; Tolmasky, M.E. Amikacin in combination with zinc pyrithione prevents growth of a carbapenem-resistant/multidrug-resistant Klebsiella pneumoniae isolate. Int. J. Antimicrob. Agents 2021, in press. [Google Scholar]
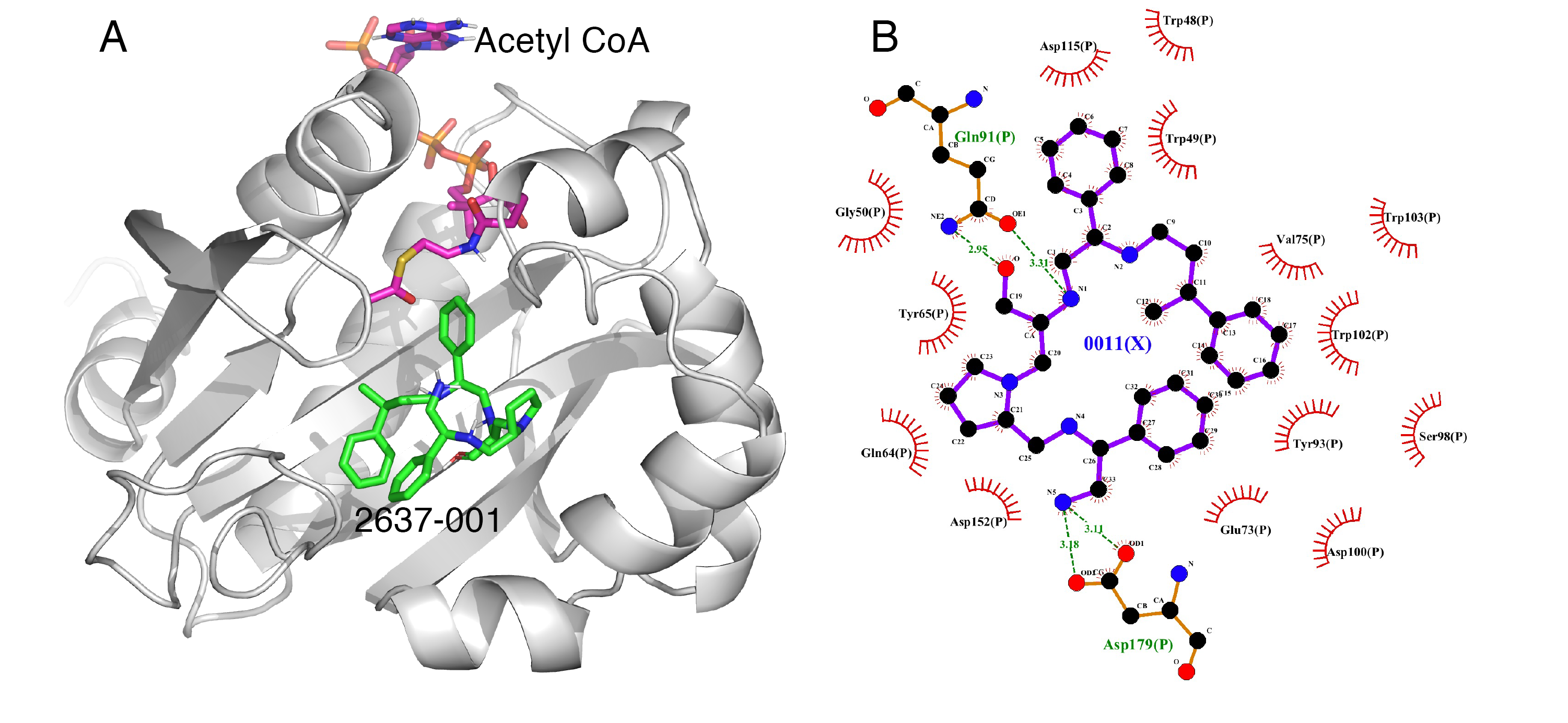




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Chemical Structure | Functionalities | %Inhibition (Average, n = 10) | Standard Error | Delta G Kcal/mL (Average, n = 3) |
|---|---|---|---|---|---|
| 2637.001 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 62 | 4 | −9.5 ± 0.1 |
| R1 analogs | |||||
| 2637.002 |  | R1: S-methyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 18 * | 2 | −8.7 ± 0.1 |
| 2637.003 | 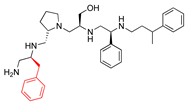 | R1: S-benzyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 20 * | 3 | −8.0 ± 0.1 |
| 2637.020 | 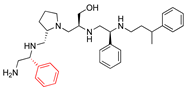 | R1: R-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 73 | 4 | −8.5 ± 0.3 |
| R2 analogs | |||||
| 2637.021 |  | R1: S-phenyl R2: R-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 28 * | 4 | −9.5 ± 0.3 |
| R3 analogs | |||||
| 2637.004 | 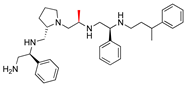 | R1: S-phenyl R2: S-pyrrolidine R3: S-methyl R4: S-phenyl R5: 3-phenylbutyl | 39 | 6 | −9.2 ± 0.1 |
| 2637.005 | 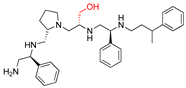 | R1: S-phenyl R2: S-pyrrolidine R3: R-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 24 * | 2 | −8.2 ± 0.1 |
| 2637.019 | 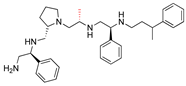 | R1: S-phenyl R2: S-pyrrolidine R3: R-methyl R4: S-phenyl R5: 3-phenylbutyl | 74 | 9 | −9.2 ± 0.1 |
| R4 analogs | |||||
| 2637.006 | 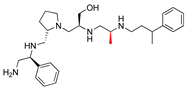 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-methyl R5: 3-phenylbutyl | 23 * | 2 | −7.9 ± 0.2 |
| 2637.022 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: R-phenyl R5: 3-phenylbutyl | 28 * | 4 | −9.6 ± 0.1 |
| R5 analogs | |||||
| 2637.007 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: ethyl | 60 | 1 | −9.5 ± 0.1 |
| 2637.008 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: butyl | 17 * | 2 | −9.4 ± 0.1 |
| 2637.010 | 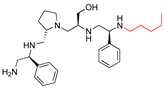 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: pentyl | 71 | 2 | −9.1 ± 0.2 |
| 2637.012 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 2-phenylbutyl | 40 | 6 | −8.5 ± 0.2 |
| 2637.011 | 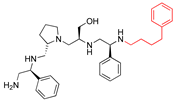 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: phenylbutyl | 66 | 6 | −8.1 ± 0.2 |
| 2637.013 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: phenylpropyl | 62 | 3 | −9.1 ± 0.1 |
| 2637.014 | 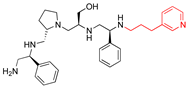 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: (pyridiin−3-yl)propyl | 46 | 3 | −9.1 ± 0.2 |
| Truncation Analogs | |||||
| 2637.015 |  | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: hydrogen | 26 * | 3 | −8.7 ± 0.1 |
| 2637.016 | 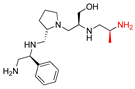 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-methyl R5: hydrogen | 20 * | 3 | −8.2 ± 0.1 |
| 2637.017 | 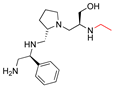 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: modified to ethyl R5: nothing | 21 * | 3 | −8.4 ± 0.1 |
| 2637.018 | 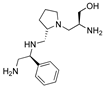 | R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: nothing R5: nothing | 17 * | 2 | −8.2 ± 0.1 |
| Compound Dose (μM) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 4 | 8 | 16 | 24 | ||||||||||||
| Compound Name | R Group Modified | IC50 μM | 95% CI | IC50 μM | 95% CI | IC50 μM | 95% CI | IC50 μM | 95% CI | IC50 μM | 95% CI | |||||
| 2637.001 | NA | 24.9 | 18.2 | 31.5 | 12.9 | 11.1 | 14.7 | 8.5 | 5.5 | 11.5 | 0.8 | 0.2 | 1.3 | 0.2 | 0.2 | 1.3 |
| 2637.020 | R1 | 23.6 | 21.2 | 26.0 | 12.6 | 10.3 | 14.8 | 9.5 | 7.5 | 11.4 | 0.3 | 0.2 | 1.2 | 0.2 | 0.1 | 0.2 |
| 2637.004 | R3 | 33.9 | 28.5 | 39.3 | 25.0 * | 18.6 | 31.3 | 20.6 * | 14.9 | 26.2 | 10.1 * | 6.5 | 13.7 | 4.8 | 1.0 | 8.6 |
| 2637.019 | R3 | 31.9 | 24.6 | 39.2 | 33.1 * | 25.6 | 40.5 | 11.5 | 9.3 | 13.7 | 0.3 | 0.3 | 0.3 | NA | NA | NA |
| 2637.007 | R5 | 29.9 | 25.2 | 34.6 | 14.8 | 10.3 | 19.3 | 8.8 | 6.9 | 10.7 | 4.0 * | 2.4 | 5.6 | 3.0 | 0.8 | 5.2 |
| 2637.010 | R5 | 31.0 | 25.6 | 36.4 | 9.2 | 7.6 | 10.8 | 5.5 | 4.1 | 6.9 | 1.7 | 0.3 | 3.3 | 1.0 | 0.2 | 1.8 |
| 2637.012 | R5 | 33.3 | 27.4 | 39.1 | 25.9 * | 21.8 | 29.9 | 20.6 * | 17.9 | 23.3 | 10.4 * | 9.1 | 11.6 | 0.3 | 0.3 | 0.3 |
| 2637.011 | R5 | 22.7 | 18.3 | 27.0 | 16.0 | 14.0 | 18.0 | 11.0 | 9.9 | 12.2 | 0.6 | 0.2 | 1.9 | 0.3 | 0.3 | 0.3 |
| 2637.013 | R5 | 29.7 | 24.2 | 35.2 | 15.3 | 12.6 | 18.1 | 8.0 | 5.9 | 10.0 | 0.7 | 0.2 | 1.7 | 0.1 | 0.2 | 0.5 |
| 2637.014 | R5 | 33.6 | 29.1 | 38.1 | 24.9 * | 20.4 | 29.4 | 19.5 * | 14.8 | 24.2 | 9.1 * | 3.6 | 14.6 | 2.8 | 0.6 | 4.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, K.; Magallon, J.; Reeves, C.; Phan, K.; Vu, P.; Oakley-Havens, C.L.; Kwan, S.; Ramirez, M.S.; LaVoi, T.; Donow, H.; et al. Inhibition of Aminoglycoside 6′-N-acetyltransferase Type Ib (AAC(6′)-Ib): Structure–Activity Relationship of Substituted Pyrrolidine Pentamine Derivatives as Inhibitors. Biomedicines 2021, 9, 1218. https://doi.org/10.3390/biomedicines9091218
Rocha K, Magallon J, Reeves C, Phan K, Vu P, Oakley-Havens CL, Kwan S, Ramirez MS, LaVoi T, Donow H, et al. Inhibition of Aminoglycoside 6′-N-acetyltransferase Type Ib (AAC(6′)-Ib): Structure–Activity Relationship of Substituted Pyrrolidine Pentamine Derivatives as Inhibitors. Biomedicines. 2021; 9(9):1218. https://doi.org/10.3390/biomedicines9091218
Chicago/Turabian StyleRocha, Kenneth, Jesus Magallon, Craig Reeves, Kimberly Phan, Peter Vu, Crista L. Oakley-Havens, Stella Kwan, Maria Soledad Ramirez, Travis LaVoi, Haley Donow, and et al. 2021. "Inhibition of Aminoglycoside 6′-N-acetyltransferase Type Ib (AAC(6′)-Ib): Structure–Activity Relationship of Substituted Pyrrolidine Pentamine Derivatives as Inhibitors" Biomedicines 9, no. 9: 1218. https://doi.org/10.3390/biomedicines9091218