
When Activator and Inhibitor of PPARα Do the Same: Consequence for Differentiation of Human Intestinal Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture and Treatment
2.2. Proliferation Assay
2.3. In-Cell ELISA (ICE)
2.4. Immunocytochemistry
2.5. Multiplex Immunofluorescence Staining
2.6. Oil Red O Staining and Quantification of Lipid Content
2.7. Immunohistochemical Detection of PPARα
2.8. Statistical Evaluation
3. Results
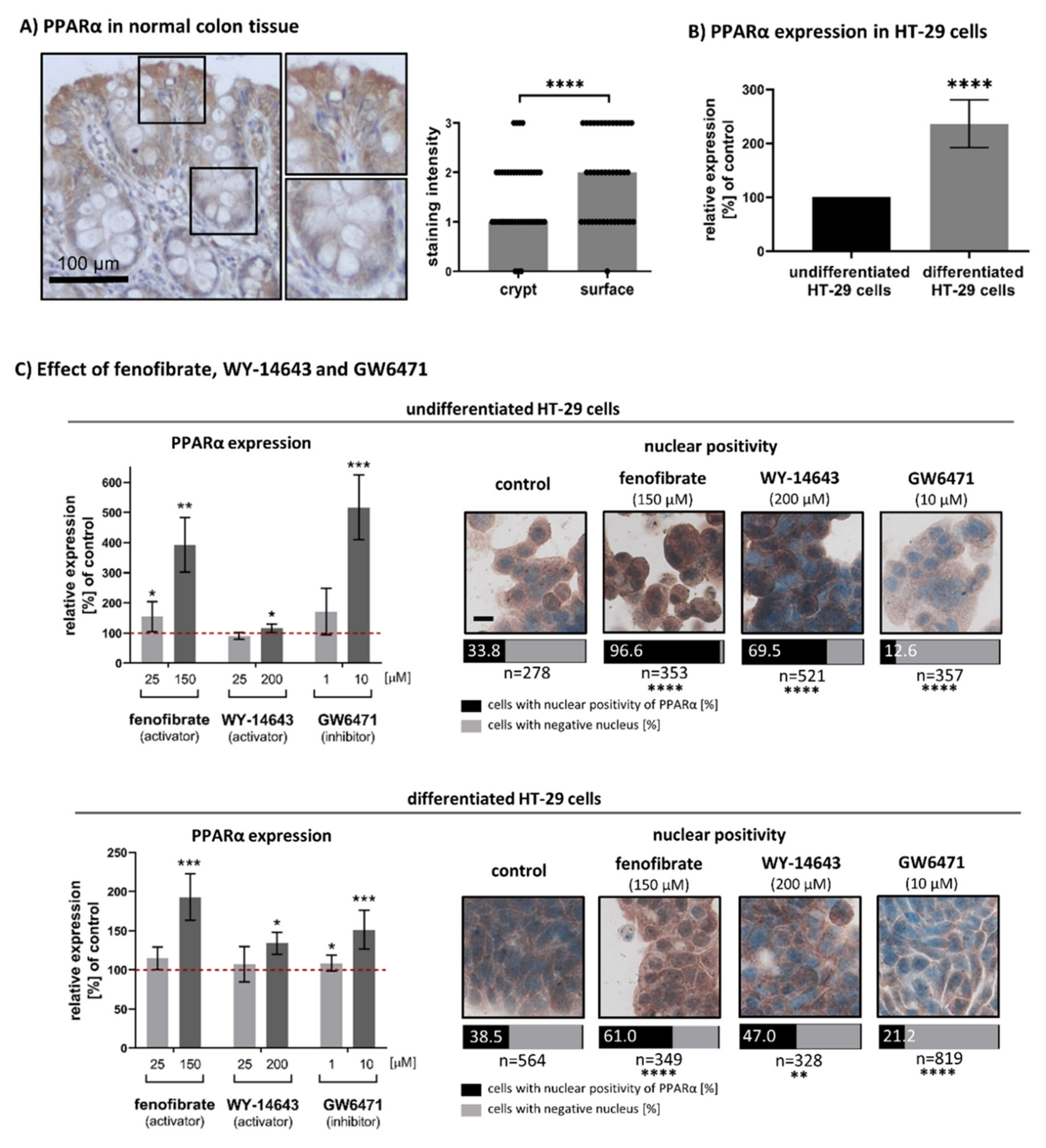
3.1. Expression and Nuclear Localisation of PPARα in Undifferentiated and Differentiated Intestinal Cells
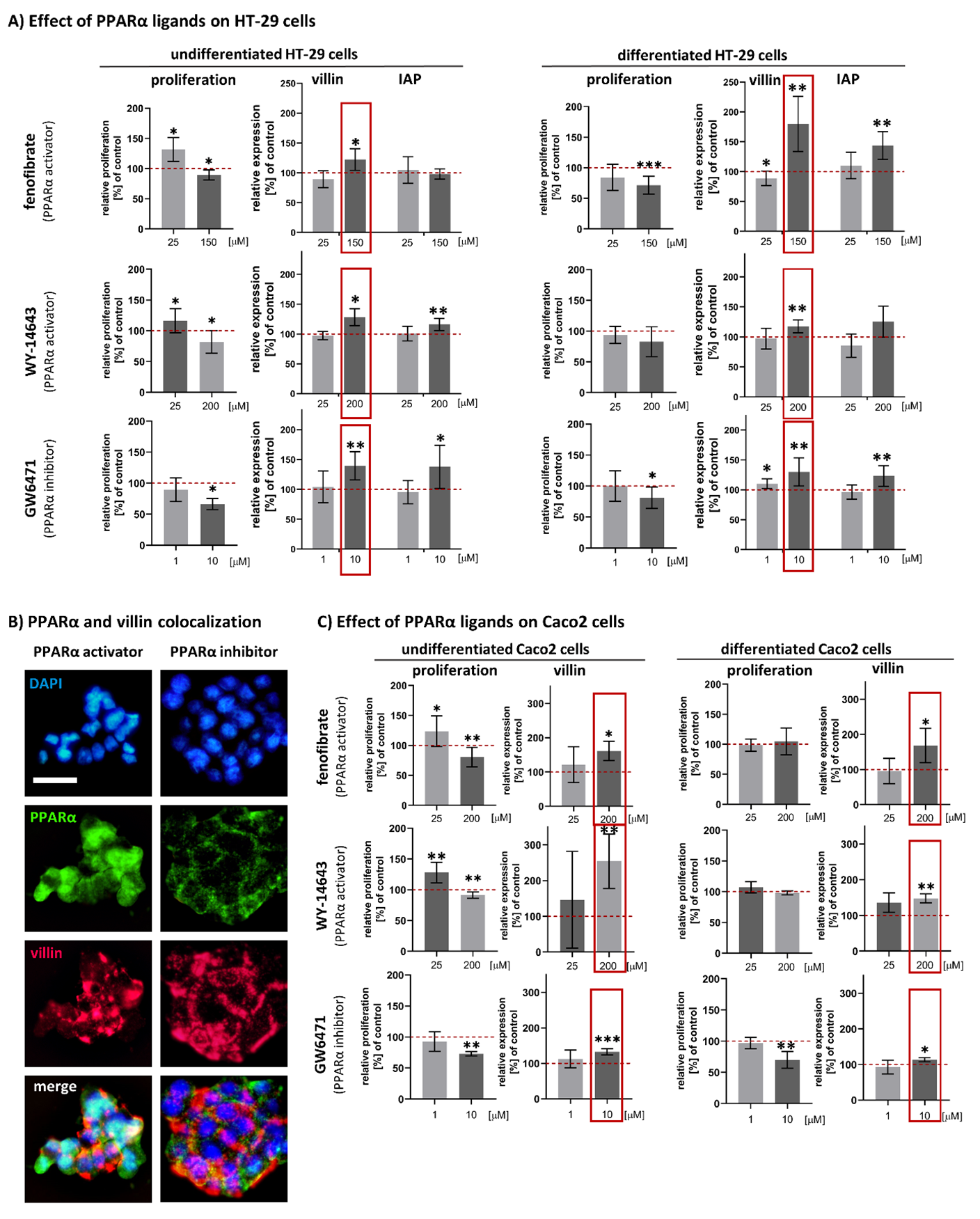
3.2. Effect of Fenofibrate, WY-14643 and GW6471 on the Cell Proliferation Activity of HT-29
3.3. Effects of Fenofibrate, WY-14643 and GW6471 on Expression of Intestinal Differentiation Markers (Villin and IAP) in HT-29 Cells
3.4. Confirmation of the Effect of Fenofibrate, WY-14643 and GW6471 on Cell Proliferation Activity and Villin Expression in Caco2 Cell Line
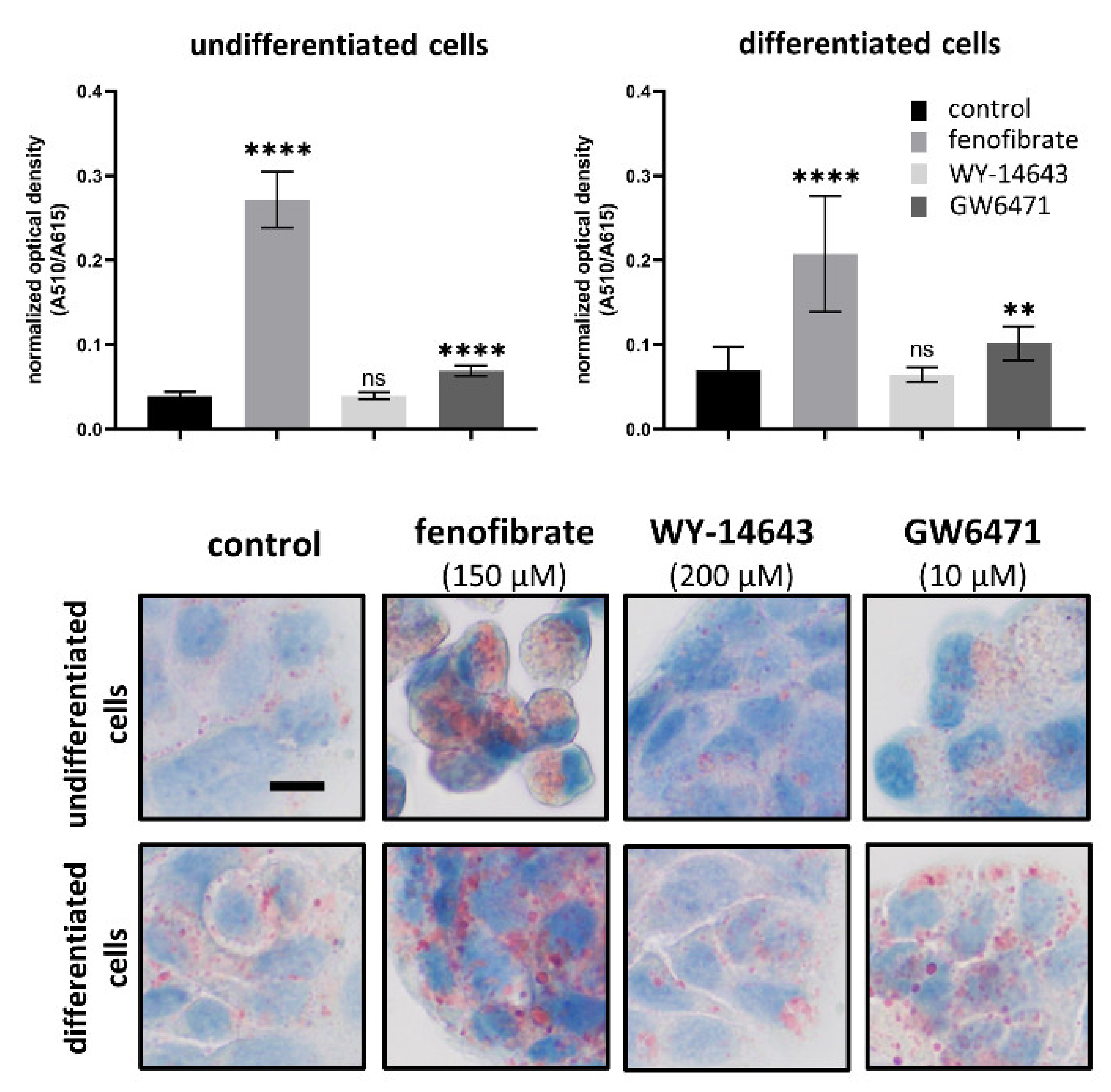
3.5. Effects of Fenofibrate, WY-14643 and GW6471 on Lipid Content
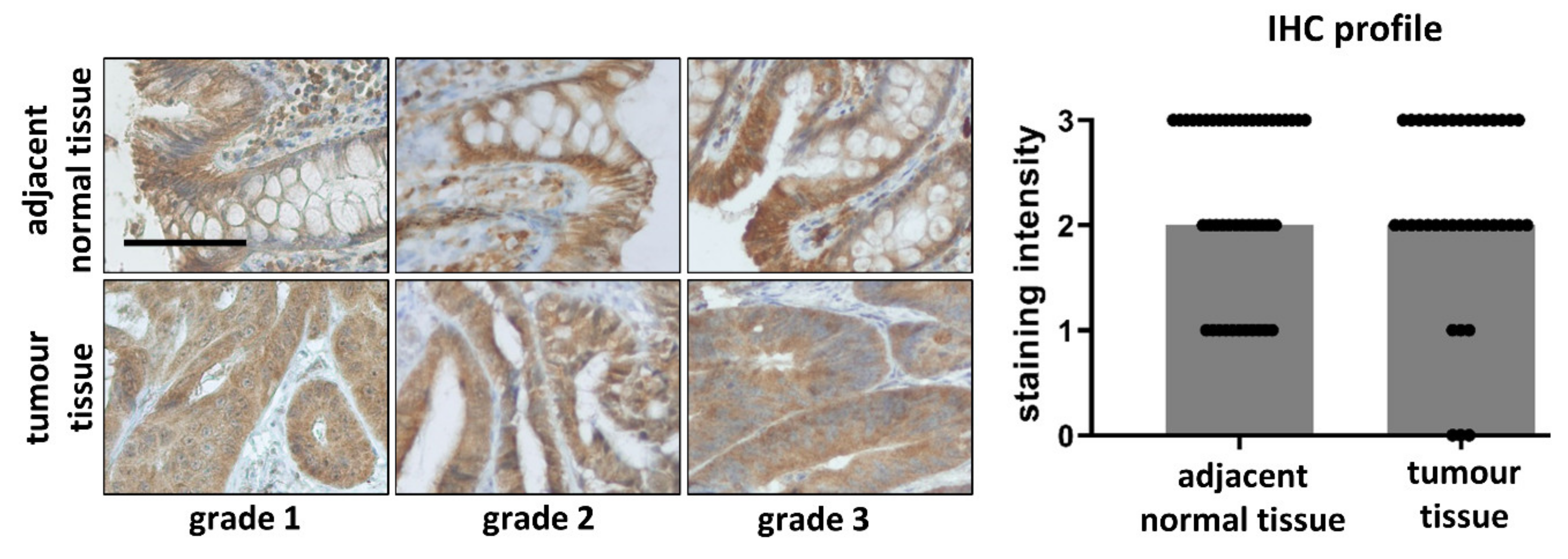
3.6. Comparison of PPARα in Tumour and Adjacent Normal Tissue Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, B.D.; Wood, C.R.; Watkins, A.M.; Das, K.P.; Lau, C.S. Peroxisome Proliferator-Activated Receptors Alpha, Beta, and Gamma mRNA and Protein Expression in Human Fetal Tissues. PPAR Res. 2010, 2010, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARα: Energy Combustion, Hypolipidemia, Inflammation and Cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Morinishi, T.; Tokuhara, Y.; Ohsaki, H.; Ibuki, E.; Kadota, K.; Hirakawa, E. Activation and Expression of Peroxisome Proliferator-Activated Receptor Alpha Are Associated with Tumorigenesis in Colorectal Carcinoma. PPAR Res. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a Key Nutritional and Environmental Sensor for Metabolic Adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuno, A.; Hirano, T.; Hayashi, T.; Mori, Y.; Yamamoto, T.; Nagashima, M.; Shiraishi, Y.; Ito, Y.; Adachi, M. The effects of statin and fibrate on lowering small dense LDL- cholesterol in hyperlipidemic patients with type 2 diabetes. J. Atheroscler. Thromb. 2007, 14, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.C.; Simes, R.J.; Barter, P.J.; Best, J.; Scott, R.A.P.; Taskinen, M.-R.; Forder, P.M.; Pillai, A.; Davis, T.M.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Absence of Functional Peroxisome Proliferator-Activated Receptor-A Enhanced Ileum Permeability During Experimental Colitis. Shock 2007, 28, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bajwa, P.J.; Carson, M.J.; Jeske, D.R.; Cong, Y.; Elson, C.O.; Lytle, C.; Straus, D.S. Fenofibrate Represses Interleukin-17 and Interferon-γ Expression and Improves Colitis in Interleukin-10–Deficient Mice. Gastroenterology 2007, 133, 108–123. [Google Scholar] [CrossRef]
- Grabacka, M.; Płonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome Proliferator–Activated Receptor α Activation Decreases Metastatic Potential of Melanoma Cells In vitro via Down-Regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [Green Version]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnés, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPAR agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Q.; Zhang, J.; Yang, G.; Shao, Z.; Luo, J.; Fan, M.; Ni, C.; Wu, Z.; Hu, X. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway. BMC Cancer 2014, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Su, C.; Jiang, M.; Shen, Y.; Shi, A.; Zhao, F.; Chen, R.; Shen, Z.; Bao, J.; Tang, W. Fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of LncRNA MEG3. Biochem. Biophys. Res. Commun. 2016, 471, 290–295. [Google Scholar] [CrossRef]
- Su, C.; Shi, A.; Cao, G.; Tao, T.; Chen, R.; Hu, Z.; Shen, Z.; Tao, H.; Cao, B.; Hu, D.; et al. Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochem. Biophys. Res. Commun. 2015, 460, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.A.; Holland, C.M.; Charnock-Jones, D.S.; Smith, S.K. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol. Cancer 2006, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Suchanek, K.M.; May, F.J.; Robinson, J.A.; Lee, W.J.; Holman, N.A.; Monteith, G.R.; Roberts-Thomson, S.J. Peroxisome proliferator-activated receptor α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol. Carcinog. 2002, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Cizkova, K.; Steigerova, J.; Gursky, J.; Ehrmann, J. Stimulating effect of normal-dosing of fibrates on cell proliferation: Word of warning. Lipids Health Dis. 2016, 15, 164. [Google Scholar] [CrossRef] [Green Version]
- Tauber, Z.; Koleckova, M.; Cizkova, K. Peroxisome proliferator-activated receptor ɑ (PPARɑ)–cytochrome P450 epoxygenases-soluble epoxide hydrolase axis in ER + PR + HER2− breast cancer. Med. Mol. Morphol. 2020, 53, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, X.-G.; Lou, Y.-J. Time-dependence of cardiomyocyte differentiation disturbed by peroxisome proliferator-activated receptor α inhibitor GW6471 in murine embryonic stem cells in vitro. Acta Pharmacol. Sin. 2007, 28, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Lee, J.-Y.; Teraminami, A.; Kim, Y.-I.; Hirai, S.; Uemura, T.; Inoue, H.; Takahashi, N.; Kawada, T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J. Lipid Res. 2011, 52, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benameur, T.; Tual-Chalot, S.; Andriantsitohaina, R.; Martínez, M.C. PPARα Is Essential for Microparticle-Induced Differentiation of Mouse Bone Marrow-Derived Endothelial Progenitor Cells and Angiogenesis. PLoS ONE 2010, 5, e12392. [Google Scholar] [CrossRef] [Green Version]
- Sharifpanah, F.; Wartenberg, M.; Hannig, M.; Piper, H.-M.; Sauer, H. Peroxisome Proliferator-Activated Receptor α Agonists Enhance Cardiomyogenesis of Mouse ES Cells by Utilization of a Reactive Oxygen Species-Dependent Mechanism. Stem Cells 2008, 26, 64–71. [Google Scholar] [CrossRef]
- Vergori, L.; Lauret, E.; Gaceb, A.; Beauvillain, C.; Andriantsitohaina, R.; Martinez, M.C. PPARα Regulates Endothelial Progenitor Cell Maturation and Myeloid Lineage Differentiation Through a NADPH Oxidase-Dependent Mechanism in Mice. Stem Cells 2015, 33, 1292–1303. [Google Scholar] [CrossRef]
- Gong, K.; Qu, B.; Wang, C.; Zhou, J.; Liao, D.; Zheng, W.; Pan, X. Peroxisome Proliferator-Activated Receptor α Facilitates Osteogenic Differentiation in MC3T3-E1 Cells via the Sirtuin 1-Dependent Signaling Pathway. Mol. Cells 2017, 40, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kömüves, L.G.; Hanley, K.; Lefebvre, A.-M.; Man, M.-Q.; Ng, D.C.; Bikle, D.D.; Williams, M.L.; Elias, P.M.; Auwerx, J.; Feingold, K.R. Stimulation of PPARα Promotes Epidermal Keratinocyte Differentiation In Vivo. J. Investig. Dermatol. 2000, 115, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Huin, C.; Corriveau, L.; Bianchi, A.; Keller, J.M.; Collet, P.; Krémarik-Bouillaud, P.; Domenjoud, L.; Bécuwe, P.; Schohn, H.; Ménard, D.; et al. Differential Expression of Peroxisome Proliferator-activated Receptors (PPARs) in the Developing Human Fetal Digestive Tract. J. Histochem. Cytochem. 2000, 48, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cizkova, K.; Rajdova, A.; Ehrmann, J. Spatio-Temporal Expression of Peroxisome Proliferator-Activated Receptor α During Human Prenatal Development. Basic Clin. Pharmacol. Toxicol. 2015, 116, 361–366. [Google Scholar] [CrossRef]
- Bünger, M.; Bosch, H.M.; Van Der Meijde, J.; Kersten, S.; Hooiveld, G.J.; Muller, M. Genome-wide analysis of PPARα activation in murine small intestine. Physiol. Genom. 2007, 30, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant Expression of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer and Their Association with Cancer Progression and Prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef]
- Huin, C.; Schohn, H.; Hatier, R.; Bentejac, M.; Antunes, L.; Plénat, F.; Bugaut, M.; Dauça, M. Expression of peroxisome proliferator-activated receptors alpha and gamma in differentiating human colon carcinoma Caco-2 cells. Biol. Cell 2002, 94, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Bourgine, J.; Billaut-Laden, I.; Happillon, M.; Lo-Guidice, J.-M.; Maunoury, V.; Imbenotte, M.; Broly, F. Gene Expression Profiling of Systems Involved in the Metabolism and the Disposition of Xenobiotics: Comparison between Human Intestinal Biopsy Samples and Colon Cell Lines. Drug Metab. Dispos. 2012, 40, 694–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cizkova, K.; Birke, P.; Malohlava, J.; Tauber, Z.; Huskova, Z.; Ehrmann, J. HT-29 and Caco2 Cell Lines Are Suitable Models for Studying the Role of Arachidonic Acid-Metabolizing Enzymes in Intestinal Cell Differentiation. Cells Tissues Organs 2019, 208, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Fujiki, Y. Ligand-dependent nucleo-cytoplasmic shuttling of peroxisome proliferator-activated receptors, PPARα and PPARγ. Genes Cells 2012, 17, 576–596. [Google Scholar] [CrossRef]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. Eur. J. Cell Biol. 2011, 90, 657–664. [Google Scholar] [CrossRef]
- Chen, L.; Peng, J.; Wang, Y.; Jiang, H.; Wang, W.; Dai, J.; Tang, M.; Wei, Y.; Kuang, H.; Xu, G.; et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal: The anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am. J. Transl. Res. 2020, 12, 428–446. [Google Scholar]
- Majeed, Y.; Upadhyay, R.; Alhousseiny, S.; Taha, T.; Musthak, A.; Shaheen, Y.; Jameel, M.; Triggle, C.R.; Ding, H. Potent and PPARα-independent anti-proliferative action of the hypolipidemic drug fenofibrate in VEGF-dependent angiosarcomas in vitro. Sci. Rep. 2019, 9, 6316. [Google Scholar] [CrossRef]
- Jan, C.-I.; Tsai, M.-H.; Chiu, C.-F.; Huang, Y.-P.; Liu, C.J.; Chang, N.W. Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. Int. J. Biol. Sci. 2016, 12, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Binello, E.; Mormone, E.; Emdad, L.; Kothari, H.; Germano, I.M. Characterization of fenofibrate-mediated anti-proliferative pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. J. Neuro-Oncol. 2014, 117, 225–234. [Google Scholar] [CrossRef]
- Jiao, H. Cytotoxic Effect of Peroxisome Proliferator Fenofibrate on Human HepG2 Hepatoma Cell Line and Relevant Mechanisms. Toxicol. Appl. Pharmacol. 2002, 185, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Holland, C.M.; Saidi, S.A.; Evans, A.L.; Sharkey, A.M.; Latimer, J.A.; Crawford, R.A.; Charnock-Jones, D.S.; Print, C.G.; Smith, S.K. Transcriptome analysis of endometrial cancer identifies peroxisome proliferator-activated receptors as potential thera-peutic targets. Mol. Cancer Ther. 2004, 3, 993–1001. [Google Scholar]
- Schmeel, L.C.; Schmeel, F.C.; Schmidt-Wolf, I.G.H. In Vitro Apoptosis Induction by Fenofibrate in Lymphoma and Multiple Myeloma. Anticancer. Res. 2017, 37, 3513–3520. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.P.; Won, S.S.; Jin, S.W.; Lee, G.H.; Pham, T.H.; Choi, J.H.; Kang, K.W.; Jeong, H.G. WY-14643 Regulates CYP1B1 Expression through Peroxisome Proliferator-Activated Receptor α-Mediated Signaling in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, F.; Morita, M.; Iwasaki, K.; Takeda, S.; Hayashi, H. Effects of WY-14643 on peroxisomal enzyme activity and hormone secretion in immortalized human trophoblast cells. Biol. Pharm. Bull. 2009, 32, 1278–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florio, R.; De Lellis, L.; DI Giacomo, V.; di Marcantonio, M.C.; Cristiano, L.; Basile, M.; Verginelli, F.; Verzilli, D.; Ammazzalorso, A.; Prasad, S.C.; et al. Effects of PPARα inhibition in head and neck paraganglioma cells. PLoS ONE 2017, 12, e0178995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARα Induces Cell Cycle Arrest and Apoptosis, and Synergizes with Glycolysis Inhibition in Kidney Cancer Cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef]
- Castelli, V.; Catanesi, M.; Alfonsetti, M.; Laezza, C.; Lombardi, F.; Cinque, B.; Cifone, M.G.; Ippoliti, R.; Benedetti, E.; Cimini, A.; et al. PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines 2021, 9, 127. [Google Scholar] [CrossRef]
- Kumar, N.; Zhao, P.; Tomar, A.; Galea, C.A.; Khurana, S. Association of Villin with Phosphatidylinositol 4,5-Bisphosphate Regulates the Actin Cytoskeleton. J. Biol. Chem. 2004, 279, 3096–3110. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.; George, S.P. Regulation of cell structure and function by actin-binding proteins: Villin’s perspective. FEBS Lett. 2008, 582, 2128–2139. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhou, Y.; Wang, X.; Chung, D.H.; Evers, B.M. Regulation of PTEN Expression in Intestinal Epithelial Cells by c-Jun NH2-Terminal Kinase Activation and Nuclear Factor-κB Inhibition. Cancer Res. 2007, 67, 7773–7781. [Google Scholar] [CrossRef] [Green Version]
- De Araujo, W.; Vidal, F.C.B.; Souza, W.; Junior, J.C.D.F.; De Souza, W.; Morgado-Diaz, J.A. PI3K/Akt and GSK-3β prevents in a differential fashion the malignant phenotype of colorectal cancer cells. J. Cancer Res. Clin. Oncol. 2010, 136, 1773–1782. [Google Scholar] [CrossRef]
- Laprise, P.; Chailler, P.; Houde, M.; Beaulieu, J.-F.; Boucher, M.-J.; Rivard, N. Phosphatidylinositol 3-Kinase Controls Human Intestinal Epithelial Cell Differentiation by Promoting Adherens Junction Assembly and p38 MAPK Activation. J. Biol. Chem. 2001, 277, 8226–8234. [Google Scholar] [CrossRef] [Green Version]
- Houde, M.; Laprise, P.; Jean, D.; Blais, M.; Asselin, C.; Rivard, N. Intestinal Epithelial Cell Differentiation Involves Activation of p38 Mitogen-activated Protein Kinase That Regulates the Homeobox Transcription Factor CDX2. J. Biol. Chem. 2001, 276, 21885–21894. [Google Scholar] [CrossRef] [Green Version]
- Banfi, C.; Auwerx, J.; Poma, F.; Tremoli, E.; Mussoni, L. Induction of plasminogen activator inhibitor 1 by the PPARα ligand, Wy-14,643, is dependent on ERK1/2 signaling pathway. Thromb. Haemost. 2003, 90, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Pauley, C.J.; Ledwith, B.J.; Kaplanski, C. Peroxisome proliferators activate growth regulatory pathways largely via peroxisome proliferator-activated receptor α-independent mechanisms. Cell. Signal. 2002, 14, 351–358. [Google Scholar] [CrossRef]
- Neuhaus, W.; Krämer, T.; Neuhoff, A.; Gölz, C.; Thal, S.C.; Förster, C.Y. Multifaceted Mechanisms of WY-14643 to Stabilize the Blood-Brain Barrier in a Model of Traumatic Brain Injury. Front. Mol. Neurosci. 2017, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leschelle, X.; Delpal, S.; Goubern, M.; Blottière, H.M.; Blachier, F. Butyrate metabolism upstream and downstream acetyl-CoA synthesis and growth control of human colon carcinoma cells. JBIC J. Biol. Inorg. Chem. 2000, 267, 6435–6442. [Google Scholar] [CrossRef] [Green Version]
- Tylichová, Z.; Slavík, J.; Ciganek, M.; Ovesná, P.; Krčmář, P.; Strakova, N.; Machala, M.; Kozubík, A.; Hofmanová, J.; Vondráček, J. Butyrate and docosahexaenoic acid interact in alterations of specific lipid classes in differentiating colon cancer cells. J. Cell. Biochem. 2018, 119, 4664–4679. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramczyk, H.; Surmacki, J.; Kopeć, M.; Olejnik, A.K.; Lubecka, K.; Fabianowska-Majewska, K. The role of lipid droplets and adipocytes in cancer. Raman imaging of cell cultures: MCF10A, MCF7, and MDA-MB-231 compared to adipocytes in cancerous human breast tissue. Analyst 2015, 140, 2224–2235. [Google Scholar] [CrossRef]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; De Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.D.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Fitchev, P.S.; Cornwell, M.L.; Greenberg, J.; Cabe, M.; Weber, C.R.; Roy, H.K.; Crawford, S.E.; Savkovic, S.D. FOXO3 Growth Inhibition of Colonic Cells Is Dependent on Intraepithelial Lipid Droplet Density. J. Biol. Chem. 2013, 288, 16274–16281. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Chen, Y.-L.; Chiang, Y.-M.; Wang, S.-G.; Lee, H.-M. Fenofibrate lowers lipid accumulation in myotubes by modulating the PPARα/AMPK/FoxO1/ATGL pathway. Biochem. Pharmacol. 2012, 84, 522–531. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Q.; Xu, C.; Cao, M.; Zhou, X.; Wang, T.; Yu, C.; Jing, F.; Chen, W.; Gao, L.; et al. Peroxisome Proliferator-Activated Receptor α Activation Induces Hepatic Steatosis, Suggesting an Adverse Effect. PLoS ONE 2014, 9, e99245. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.-W.; Wu, C.-T.; Chen, D.-R.; Yeh, C.-Y.; Lin, C. High levels of arachidonic acid and peroxisome proliferator-activated receptor-alpha in breast cancer tissues are associated with promoting cancer cell proliferation. J. Nutr. Biochem. 2013, 24, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Zuo, N.; Zheng, X.; Liu, H.; Ma, X. Fenofibrate, a PPARα agonist, protect proximal tubular cells from albumin-bound fatty acids induced apoptosis via the activation of NF-kB. Int. J. Clin. Exp. Pathol. 2015, 8, 10653–10661. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cizkova, K.; Foltynkova, T.; Hanyk, J.; Kamencak, Z.; Tauber, Z. When Activator and Inhibitor of PPARα Do the Same: Consequence for Differentiation of Human Intestinal Cells. Biomedicines 2021, 9, 1255. https://doi.org/10.3390/biomedicines9091255
Cizkova K, Foltynkova T, Hanyk J, Kamencak Z, Tauber Z. When Activator and Inhibitor of PPARα Do the Same: Consequence for Differentiation of Human Intestinal Cells. Biomedicines. 2021; 9(9):1255. https://doi.org/10.3390/biomedicines9091255
Chicago/Turabian StyleCizkova, Katerina, Tereza Foltynkova, Jiri Hanyk, Zbynek Kamencak, and Zdenek Tauber. 2021. "When Activator and Inhibitor of PPARα Do the Same: Consequence for Differentiation of Human Intestinal Cells" Biomedicines 9, no. 9: 1255. https://doi.org/10.3390/biomedicines9091255