
Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability


Abstract
:1. Introduction
2. Genetic and Metabolic Investigations in Children with GDD/ID
2.1. Chromosomal Microarray
2.2. Exome Sequencing (ES) and Comprehensive GDD/ID Gene Panels
2.3. Genome Sequencing
2.4. Fragile X Syndrome Testing
2.5. Metabolic/Biochemical Screening for Inherited Metabolic Diseases
3. Common CNVs in Non-Syndromic GDD/ID

{kind=link}
| Chromosome Region | Deletion or Duplication | Main Clinical Features | Candidate Genes | References |
|---|---|---|---|---|
| 15q11.2 | Deletion/Duplication | ID, schizophrenia, epilepsy | TUBGCP5, CYFIP1, NIPA2, NIPA1 | [74,75,76,77,78,79,80] |
| 15q13.3 | Deletion | ID, epilepsy, schizophrenia, ASD | CHRNA7 | [74,81,82,83,84,85,86] |
| 16p11.2 (distal) | Deletion | ID, ASD, obesity, schizophrenia | SH2B1 | [87,88] |
| 16p11.2 (proximal) | Deletion/Duplication | ID, language delay, ASD, obesity | MVP, CDIPT1, SEZ6L2, ASPHD1, KCTD13 | [89,90,91,92,93,94,95,96] |
| 16p12 | Deletion | Intellectual disability | UQCRC2, EEF2K, POLR3E, CDR2 | [41,97] |
| Xq28 | Duplication | Males: hypotonia, severe GDD and ID, progressive spasticity, seizures, ASD Females: milder phenotype | RAB39B, CLIC2, IRAK1, MECP2, GDI1 | [41,98,99,100,101] |
4. Common Pathways Underlying NDDs
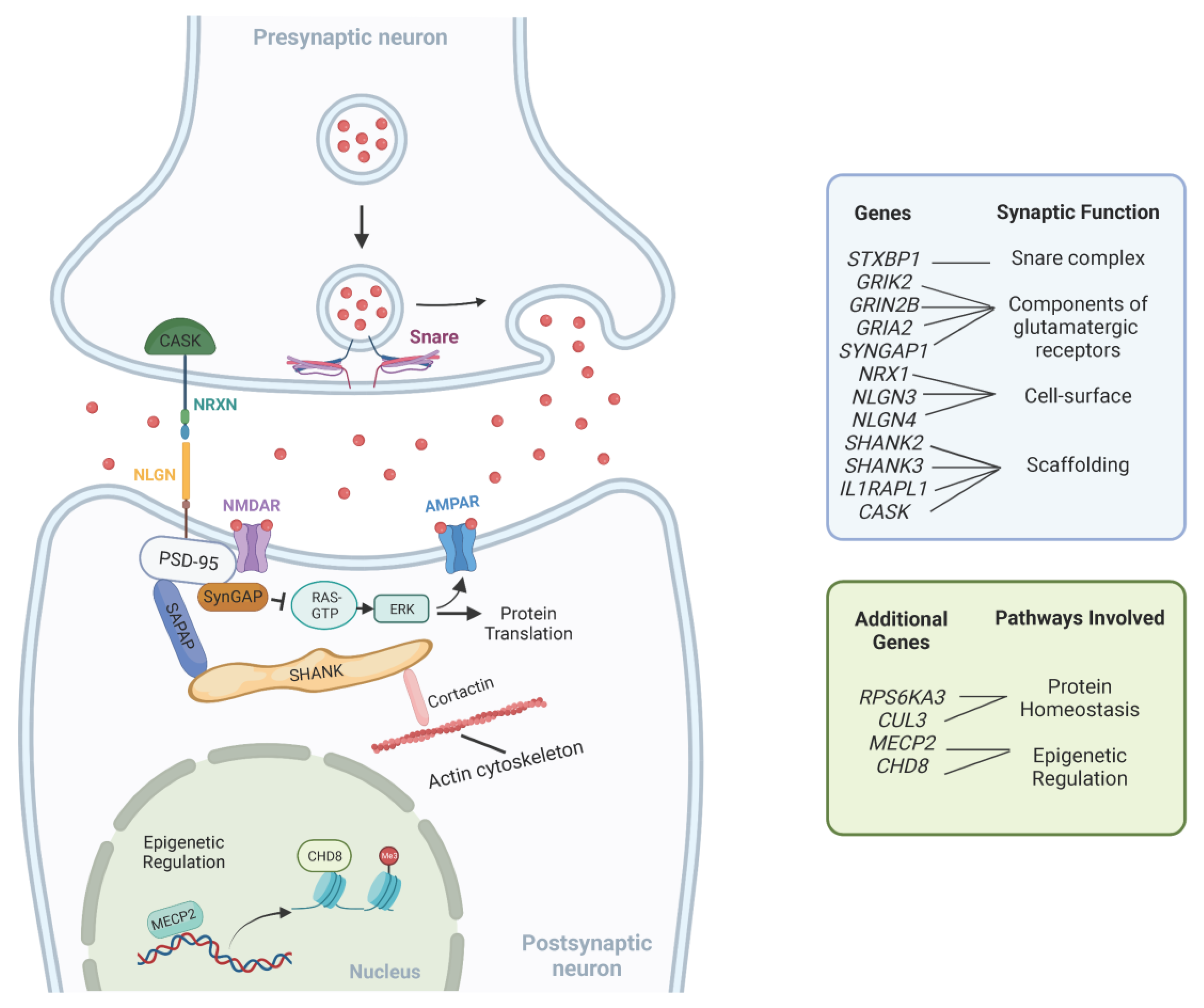
4.1. Synaptic Signaling Dysregulation
4.2. Protein Homeostasis
4.3. Epigenetic Regulation
4.4. Thalamic and Peripheral Circuits
5. Diagnostic Approach to the Evaluation of Children with Non-Syndromic GDD/ID
6. Overview of Management Principles for Children with GDD/ID
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diagnostic and Statistical Manual of Mental Disorders DSM-5. Available online: https://doi.org/10.1176/appi.books.9780890425596 (accessed on 29 November 2022).
- Michelson, D.J.; Clark, R.D. Optimizing Genetic Diagnosis of Neurodevelopmental Disorders in the Clinical Setting. Clin. Lab. Med. 2020, 40, 231–256. [Google Scholar] [CrossRef]
- Kaufman, L.; Ayub, M.; Vincent, J.B. The Genetic Basis of Non-Syndromic Intellectual Disability: A Review. J. Neurodev. Disord. 2010, 2, 182–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurazzi, P.; Pirozzi, F. Advances in Understanding—Genetic Basis of Intellectual Disability. F1000Research 2016, 5, 599. [Google Scholar] [CrossRef]
- Nair, R.; Chen, M.; Dutt, A.S.; Hagopian, L.; Singh, A.; Du, M. Significant Regional Inequalities in the Prevalence of Intellectual Disability and Trends from 1990 to 2019: A Systematic Analysis of GBD 2019. Epidemiol. Psychiatr. Sci. 2022, 31, e91. [Google Scholar] [CrossRef] [PubMed]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.W.; Kleefstra, T.; Kramer, J.M.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenberg, A.; Cederlöf, M.; McMillan, A.; Trzaskowski, M.; Kapra, O.; Fruchter, E.; Ginat, K.; Davidson, M.; Weiser, M.; Larsson, H.; et al. Discontinuity in the Genetic and Environmental Causes of the Intellectual Disability Spectrum. Proc. Natl. Acad. Sci. USA 2016, 113, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Modabbernia, A.; Mollon, J.; Boffetta, P.; Reichenberg, A. Impaired Gas Exchange at Birth and Risk of Intellectual Disability and Autism: A Meta-Analysis. J. Autism. Dev. Disord. 2016, 46, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- McDermott, S.; Bao, W.; Tong, X.; Cai, B.; Lawson, A.; Aelion, C.M. Are Different Soil Metals near the Homes of Pregnant Women Associated with Mild and Severe Intellectual Disability in Children? Dev. Med. Child Neurol. 2014, 56, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, S.A.; Caron, J. Evaluation of the Child with Global Developmental Delay and Intellectual Disability. Paediatr. Child Health 2018, 23, 403–419. [Google Scholar] [CrossRef]
- Jimenez-Gomez, A.; Standridge, S.M. A Refined Approach to Evaluating Global Developmental Delay for the International Medical Community. Pediatr. Neurol. 2014, 51, 198–206. [Google Scholar] [CrossRef]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of Intellectual Disability and Autism Comorbidity through Gene Panel Sequencing. Hum. Mutat. 2019, 40, 1346–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.; Jang, J.H.; Park, J.; Lee, I.G. Targeted Next-Generation Sequencing of Korean Patients With Developmental Delay and/or Intellectual Disability. Front. Pediatr. 2018, 6, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.; Race, V.; Keldermans, L.; Bauters, M.; Van Esch, H. Challenges in Molecular Diagnosis of X-Linked Intellectual Disability. Br. Med. Bull. 2020, 133, 36–48. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Prevalence and Architecture of de Novo Mutations in Developmental Disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef] [Green Version]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping Autosomal Recessive Intellectual Disability: Combined Microarray and Exome Sequencing Identifies 26 Novel Candidate Genes in 192 Consanguineous Families. Mol. Psychiatry 2018, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep Sequencing Reveals 50 Novel Genes for Recessive Cognitive Disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef]
- Anazi, S.; Maddirevula, S.; Faqeih, E.; Alsedairy, H.; Alzahrani, F.; Shamseldin, H.E.; Patel, N.; Hashem, M.; Ibrahim, N.; Abdulwahab, F.; et al. Clinical Genomics Expands the Morbid Genome of Intellectual Disability and Offers a High Diagnostic Yield. Mol. Psychiatry 2017, 22, 615–624. [Google Scholar] [CrossRef]
- Tan, T.Y.; Dillon, O.J.; Stark, Z.; Schofield, D.; Alam, K.; Shrestha, R.; Chong, B.; Phelan, D.; Brett, G.R.; Creed, E.; et al. Diagnostic Impact and Cost-Effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr. 2017, 171, 855–862. [Google Scholar] [CrossRef]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M.; et al. Exome and Genome Sequencing for Pediatric Patients with Congenital Anomalies or Intellectual Disability: An Evidence-Based Clinical Guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.; Hudgins, L. Professional Practice and Guidelines Committee Array-Based Technology and Recommendations for Utilization in Medical Genetics Practice for Detection of Chromosomal Abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelson, D.J.; Shevell, M.I.; Sherr, E.H.; Moeschler, J.B.; Gropman, A.L.; Ashwal, S. Evidence Report: Genetic and Metabolic Testing on Children with Global Developmental Delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2011, 77, 1629–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeschler, J.B.; Shevell, M. Committee on Genetics Comprehensive Evaluation of the Child with Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhle, R.A.; Reed, H.E.; Vo, L.C.; Mehta, S.; McGuire, K.; Veenstra-VanderWeele, J.; Pedapati, E. Clinical Diagnostic Genetic Testing for Individuals With Developmental Disorders. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 910–913. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.M.; Stockler, S. Treatable Inborn Errors of Metabolism Causing Intellectual Disability: A Systematic Literature Review. Mol. Genet. Metab. 2012, 105, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Hoytema van Konijnenburg, E.M.M.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D.M. Treatable Inherited Metabolic Disorders Causing Intellectual Disability: 2021 Review and Digital App. Orphanet J. Rare Dis. 2021, 16, 170. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-Analysis and Multidisciplinary Consensus Statement: Exome Sequencing Is a First-Tier Clinical Diagnostic Test for Individuals with Neurodevelopmental Disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [Green Version]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Waggoner, D.; Wain, K.E.; Dubuc, A.M.; Conlin, L.; Hickey, S.E.; Lamb, A.N.; Martin, C.L.; Morton, C.C.; Rasmussen, K.; Schuette, J.L.; et al. Yield of Additional Genetic Testing after Chromosomal Microarray for Diagnosis of Neurodevelopmental Disability and Congenital Anomalies: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 1105–1113. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Lupo, P.J.; Stankiewicz, P.; Schaaf, C.P. An Estimation of the Prevalence of Genomic Disorders Using Chromosomal Microarray Data. J. Hum. Genet. 2018, 63, 795–801. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of Penetrance for Recurrent Pathogenic Copy-Number Variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef]
- Smajlagić, D.; Lavrichenko, K.; Berland, S.; Helgeland, Ø.; Knudsen, G.P.; Vaudel, M.; Haavik, J.; Knappskog, P.M.; Njølstad, P.R.; Houge, G.; et al. Population Prevalence and Inheritance Pattern of Recurrent CNVs Associated with Neurodevelopmental Disorders in 12,252 Newborns and Their Parents. Eur. J. Hum. Genet. 2021, 29, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.; Kim, Y.; Han, E.; Park, J.; Chae, H.; Kwon, A.; Choi, H.; Kim, J.; Son, J.O.; Lee, S.J.; et al. Chromosomal Microarray Analysis as a First-Tier Clinical Diagnostic Test in Patients With Developmental Delay/Intellectual Disability, Autism Spectrum Disorders, and Multiple Congenital Anomalies: A Prospective Multicenter Study in Korea. Ann. Lab. Med. 2019, 39, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.W.; Chan, K.Y.K.; Leung, K.K.P.; Au, P.K.C.; Tam, W.-K.; Li, S.K.M.; Luk, H.-M.; Kan, A.S.Y.; Chung, B.H.Y.; Lo, I.F.M.; et al. Experience of Chromosomal Microarray Applied in Prenatal and Postnatal Settings in Hong Kong. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Doccini, V.; Bernardini, L.; Novelli, A.; Loddo, S.; Capalbo, A.; Filippi, T.; Carey, J.C. Confirmation of Chromosomal Microarray as a First-Tier Clinical Diagnostic Test for Individuals with Developmental Delay, Intellectual Disability, Autism Spectrum Disorders and Dysmorphic Features. Eur. J. Paediatr. Neurol. 2013, 17, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, M.; Wiśniowiecka-Kowalnik, B.; Nowakowska, B.; Smyk, M.; Kędzior, M.; Sobecka, K.; Kutkowska-Kaźmierczak, A.; Klapecki, J.; Szczałuba, K.; Castañeda, J.; et al. The Usefulness of Array Comparative Genomic Hybridization in Clinical Diagnostics of Intellectual Disability in Children. Dev. Period Med. 2014, 18, 307–317. [Google Scholar]
- Fan, Y.; Wu, Y.; Wang, L.; Wang, Y.; Gong, Z.; Qiu, W.; Wang, J.; Zhang, H.; Ji, X.; Ye, J.; et al. Chromosomal Microarray Analysis in Developmental Delay and Intellectual Disability with Comorbid Conditions. BMC Med. Genom. 2018, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- D’Arrigo, S.; Gavazzi, F.; Alfei, E.; Zuffardi, O.; Montomoli, C.; Corso, B.; Buzzi, E.; Sciacca, F.L.; Bulgheroni, S.; Riva, D.; et al. The Diagnostic Yield of Array Comparative Genomic Hybridization Is High Regardless of Severity of Intellectual Disability/Developmental Delay in Children. J. Child Neurol. 2016, 31, 691–699. [Google Scholar] [CrossRef]
- Oğuz, S.; Arslan, U.E.; Kiper, P.Ö.Ş.; Alikaşifoğlu, M.; Boduroğlu, K.; Utine, G.E. Diagnostic Yield of Microarrays in Individuals with Non-Syndromic Developmental Delay and Intellectual Disability. J. Intellect. Disabil. Res. 2021, 65, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D.; Quality Standards Subcommittee of the American Academy of Neurology; et al. Practice Parameter: Evaluation of the Child with Global Developmental Delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Corominas, J.; Smeekens, S.P.; Nelen, M.R.; Yntema, H.G.; Kamsteeg, E.-J.; Pfundt, R.; Gilissen, C. Clinical Exome Sequencing-Mistakes and Caveats. Hum. Mutat. 2022, 43, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, D.; Heeley, J.; Vineyard, M.; Manwaring, L.; Toler, T.L.; Fassi, E.; Fiala, E.; Brown, S.; Goss, C.W.; Willing, M.; et al. The Exome Clinic and the Role of Medical Genetics Expertise in the Interpretation of Exome Sequencing Results. Genet. Med. 2017, 19, 1040–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, O.J.; Lunke, S.; Stark, Z.; Yeung, A.; Thorne, N.; Melbourne Genomics Health Alliance; Gaff, C.; White, S.M.; Tan, T.Y. Exome Sequencing Has Higher Diagnostic Yield Compared to Simulated Disease-Specific Panels in Children with Suspected Monogenic Disorders. Eur. J. Hum. Genet. 2018, 26, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Gieldon, L.; Mackenroth, L.; Kahlert, A.-K.; Lemke, J.R.; Porrmann, J.; Schallner, J.; von der Hagen, M.; Markus, S.; Weidensee, S.; Novotna, B.; et al. Diagnostic Value of Partial Exome Sequencing in Developmental Disorders. PLoS ONE 2018, 13, e0201041. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.; Carlson, M. Whole Exome Sequencing in Pediatric Neurology Patients: Clinical Implications and Estimated Cost Analysis. J. Child Neurol. 2016, 31, 887–894. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Palmer, E.E.; Schofield, D.; Shrestha, R.; Kandula, T.; Macintosh, R.; Lawson, J.A.; Andrews, I.; Sampaio, H.; Johnson, A.M.; Farrar, M.A.; et al. Integrating Exome Sequencing into a Diagnostic Pathway for Epileptic Encephalopathy: Evidence of Clinical Utility and Cost Effectiveness. Mol. Genet. Genomic. Med. 2018, 6, 186–199. [Google Scholar] [CrossRef]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic Reanalysis of Clinical Exome Data Yields Additional Diagnoses: Implications for Providers. Genet. Med. 2017, 19, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Al-Nabhani, M.; Al-Rashdi, S.; Al-Murshedi, F.; Al-Kindi, A.; Al-Thihli, K.; Al-Saegh, A.; Al-Futaisi, A.; Al-Mamari, W.; Zadjali, F.; Al-Maawali, A. Reanalysis of Exome Sequencing Data of Intellectual Disability Samples: Yields and Benefits. Clin. Genet. 2018, 94, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Basel-Salmon, L.; Orenstein, N.; Markus-Bustani, K.; Ruhrman-Shahar, N.; Kilim, Y.; Magal, N.; Hubshman, M.W.; Bazak, L. Improved Diagnostics by Exome Sequencing Following Raw Data Reevaluation by Clinical Geneticists Involved in the Medical Care of the Individuals Tested. Genet. Med. 2019, 21, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Zhang, Z.; Shi, P.; Martin, D.M.; Kong, X. Incorporation of Exome-Based CNV Analysis Makes Trio-WES a More Powerful Tool for Clinical Diagnosis in Neurodevelopmental Disorders: A Retrospective Study. Hum. Mutat. 2021, 42, 990–1004. [Google Scholar] [CrossRef]
- Xiang, J.; Ding, Y.; Yang, F.; Gao, A.; Zhang, W.; Tang, H.; Mao, J.; He, Q.; Zhang, Q.; Wang, T. Genetic Analysis of Children With Unexplained Developmental Delay and/or Intellectual Disability by Whole-Exome Sequencing. Front. Genet 2021, 12, 738561. [Google Scholar] [CrossRef] [PubMed]
- Martínez, F.; Caro-Llopis, A.; Roselló, M.; Oltra, S.; Mayo, S.; Monfort, S.; Orellana, C. High Diagnostic Yield of Syndromic Intellectual Disability by Targeted Next-Generation Sequencing. J. Med. Genet. 2017, 54, 87–92. [Google Scholar] [CrossRef]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.-I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Pekeles, H.; Accogli, A.; Boudrahem-Addour, N.; Russell, L.; Parente, F.; Srour, M. Diagnostic Yield of Intellectual Disability Gene Panels. Pediatr. Neurol. 2019, 92, 32–36. [Google Scholar] [CrossRef]
- Chérot, E.; Keren, B.; Dubourg, C.; Carré, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using Medical Exome Sequencing to Identify the Causes of Neurodevelopmental Disorders: Experience of 2 Clinical Units and 216 Patients. Clin. Genet. 2018, 93, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Snoeijen-Schouwenaars, F.M.; van Ool, J.S.; Verhoeven, J.S.; van Mierlo, P.; Braakman, H.M.H.; Smeets, E.E.; Nicolai, J.; Schoots, J.; Teunissen, M.W.A.; Rouhl, R.P.W.; et al. Diagnostic Exome Sequencing in 100 Consecutive Patients with Both Epilepsy and Intellectual Disability. Epilepsia 2019, 60, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Bowling, K.M.; Thompson, M.L.; Amaral, M.D.; Finnila, C.R.; Hiatt, S.M.; Engel, K.L.; Cochran, J.N.; Brothers, K.B.; East, K.M.; Gray, D.E.; et al. Genomic Diagnosis for Children with Intellectual Disability and/or Developmental Delay. Genome. Med. 2017, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Ewans, L.J.; Schofield, D.; Shrestha, R.; Zhu, Y.; Gayevskiy, V.; Ying, K.; Walsh, C.; Lee, E.; Kirk, E.P.; Colley, A.; et al. Whole-Exome Sequencing Reanalysis at 12 Months Boosts Diagnosis and Is Cost-Effective When Applied Early in Mendelian Disorders. Genet. Med. 2018, 20, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Ontario Health (Quality). Genome-Wide Sequencing for Unexplained Developmental Disabilities or Multiple Congenital Anomalies: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2020, 20, 1–178. [Google Scholar]
- Sun, Y.; Peng, J.; Liang, D.; Ye, X.; Xu, N.; Chen, L.; Yan, D.; Zhang, H.; Xiao, B.; Qiu, W.; et al. Genome Sequencing Demonstrates High Diagnostic Yield in Children with Undiagnosed Global Developmental Delay/Intellectual Disability: A Prospective Study. Hum. Mutat. 2022, 43, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Borch, L.A.; Parboosingh, J.; Thomas, M.A.; Veale, P. Re-Evaluating the First-Tier Status of Fragile X Testing in Neurodevelopmental Disorders. Genet. Med. 2020, 22, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Christofolini, D.M.; Abbud, E.M.; Lipay, M.V.N.; Costa, S.S.; Vianna-Morgante, A.M.; Bellucco, F.T.S.; Nogueira, S.I.; Kulikowski, L.D.; Brunoni, D.; Juliano, Y.; et al. Evaluation of Clinical Checklists for Fragile X Syndrome Screening in Brazilian Intellectually Disabled Males: Proposal for a New Screening Tool. J. Intellect. Disabil. 2009, 13, 239–248. [Google Scholar] [CrossRef]
- Giangreco, C.A.; Steele, M.W.; Aston, C.E.; Cummins, J.H.; Wenger, S.L. A Simplified Six-Item Checklist for Screening for Fragile X Syndrome in the Pediatric Population. J. Pediatr. 1996, 129, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Lubala, T.K.; Lumaka, A.; Kanteng, G.; Mutesa, L.; Mukuku, O.; Wembonyama, S.; Hagerman, R.; Luboya, O.N.; Lukusa Tshilobo, P. Fragile X Checklists: A Meta-analysis and Development of a Simplified Universal Clinical Checklist. Mol. Genet. Genomic. Med. 2018, 6, 526–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice and Guidelines Committee Clinical Genetics Evaluation in Identifying the Etiology of Autism Spectrum Disorders: 2013 Guideline Revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Vallance, H.; Sinclair, G.; Rakic, B.; Stockler-Ipsiroglu, S. Diagnostic Yield from Routine Metabolic Screening Tests in Evaluation of Global Developmental Delay and Intellectual Disability. Paediatr. Child Health 2021, 26, 344–348. [Google Scholar] [CrossRef]
- Sempere, A.; Arias, A.; Farré, G.; García-Villoria, J.; Rodríguez-Pombo, P.; Desviat, L.R.; Merinero, B.; García-Cazorla, A.; Vilaseca, M.A.; Ribes, A.; et al. Study of Inborn Errors of Metabolism in Urine from Patients with Unexplained Mental Retardation. J. Inherit. Metab. Dis. 2010, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Rosenfeld, J.A.; Shur, N.; Slavotinek, A.M.; Cox, V.A.; Hennekam, R.C.; Firth, H.V.; Willatt, L.; Wheeler, P.; Morrow, E.M.; et al. Further Clinical and Molecular Delineation of the 15q24 Microdeletion Syndrome. J. Med. Genet. 2012, 49, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large Recurrent Microdeletions Associated with Schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kovel, C.G.F.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; von Spiczak, S.; Ostertag, P.; et al. Recurrent Microdeletions at 15q11.2 and 16p13.11 Predispose to Idiopathic Generalized Epilepsies. Brain 2010, 133, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mefford, H.C.; Cooper, G.M.; Zerr, T.; Smith, J.D.; Baker, C.; Shafer, N.; Thorland, E.C.; Skinner, C.; Schwartz, C.E.; Nickerson, D.A.; et al. A Method for Rapid, Targeted CNV Genotyping Identifies Rare Variants Associated with Neurocognitive Disease. Genome. Res. 2009, 19, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/Microduplication of Proximal 15q11.2 between BP1 and BP2: A Susceptibility Region for Neurological Dysfunction Including Developmental and Language Delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.; Sikkema-Raddatz, B.; Ruijvenkamp, C.A.L.; Dijkhuizen, T.; Bijlsma, E.K.; Gijsbers, A.C.J.; Hilhorst-Hofstee, Y.; Hordijk, R.; Verbruggen, K.T.; Kerstjens-Frederikse, W.S.M.; et al. Nine Patients with a Microdeletion 15q11.2 between Breakpoints 1 and 2 of the Prader-Willi Critical Region, Possibly Associated with Behavioural Disturbances. Eur. J. Med. Genet. 2009, 52, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.K.; Nygren, A.O.H.; El Shakankiry, H.M.; Schouten, J.P.; Al Khayat, A.I.; Ridha, A.; Al Ali, M.T. Detection of a Novel Familial Deletion of Four Genes between BP1 and BP2 of the Prader-Willi/Angelman Syndrome Critical Region by Oligo-Array CGH in a Child with Neurological Disorder and Speech Impairment. Cytogenet. Genome. Res. 2007, 116, 135–140. [Google Scholar] [CrossRef] [PubMed]
- von der Lippe, C.; Rustad, C.; Heimdal, K.; Rødningen, O.K. 15q11.2 Microdeletion—Seven New Patients with Delayed Development and/or Behavioural Problems. Eur. J. Med. Genet. 2011, 54, 357–360. [Google Scholar] [CrossRef]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; de Kovel, C.; Baker, C.; von Spiczak, S.; et al. 15q13.3 Microdeletions Increase Risk of Idiopathic Generalized Epilepsy. Nat. Genet. 2009, 41, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; Stevenson, R.E.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A Recurrent 15q13.3 Microdeletion Syndrome Associated with Mental Retardation and Seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef] [PubMed]
- van Bon, B.W.M.; Mefford, H.C.; Menten, B.; Koolen, D.A.; Sharp, A.J.; Nillesen, W.M.; Innis, J.W.; de Ravel, T.J.L.; Mercer, C.L.; Fichera, M.; et al. Further Delineation of the 15q13 Microdeletion and Duplication Syndromes: A Clinical Spectrum Varying from Non-Pathogenic to a Severe Outcome. J. Med. Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shachar, S.; Lanpher, B.; German, J.R.; Qasaymeh, M.; Potocki, L.; Nagamani, S.C.S.; Franco, L.M.; Malphrus, A.; Bottenfield, G.W.; Spence, J.E.; et al. Microdeletion 15q13.3: A Locus with Incomplete Penetrance for Autism, Mental Retardation, and Psychiatric Disorders. J. Med. Genet. 2009, 46, 382–388. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Wing, K.; Sadighi Akha, E.; Knight, S.J.L.; Bölte, S.; Schmötzer, G.; Duketis, E.; Poustka, F.; Klauck, S.M.; Poustka, A.; et al. A 15q13.3 Microdeletion Segregating with Autism. Eur. J. Hum. Genet. 2009, 17, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J.; et al. Microdeletion/Duplication at 15q13.2q13.3 among Individuals with Features of Autism and Other Neuropsychiatric Disorders. J. Med. Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann-Gagescu, R.; Mefford, H.C.; Cowan, C.; Glew, G.M.; Hing, A.V.; Wallace, S.; Bader, P.I.; Hamati, A.; Reitnauer, P.J.; Smith, R.; et al. Recurrent 200-Kb Deletions of 16p11.2 That Include the SH2B1 Gene Are Associated with Developmental Delay and Obesity. Genet. Med. 2010, 12, 641–647. [Google Scholar] [CrossRef] [Green Version]
- Bochukova, E.G.; Huang, N.; Keogh, J.; Henning, E.; Purmann, C.; Blaszczyk, K.; Saeed, S.; Hamilton-Shield, J.; Clayton-Smith, J.; O’Rahilly, S.; et al. Large, Rare Chromosomal Deletions Associated with Severe Early-Onset Obesity. Nature 2010, 463, 666–670. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.R.; Green, T.; et al. Association between Microdeletion and Microduplication at 16p11.2 and Autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.; Novelli, A.; Bernardini, L.; Igliozzi, R.; Parrini, B. Further Characterization of the New Microdeletion Syndrome of 16p11.2-P12.2. Am. J. Med. Genet. A 2009, 149A, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, E.K.; Gijsbers, A.C.J.; Schuurs-Hoeijmakers, J.H.M.; van Haeringen, A.; Fransen van de Putte, D.E.; Anderlid, B.-M.; Lundin, J.; Lapunzina, P.; Pérez Jurado, L.A.; Delle Chiaie, B.; et al. Extending the Phenotype of Recurrent Rearrangements of 16p11.2: Deletions in Mentally Retarded Patients without Autism and in Normal Individuals. Eur. J. Med. Genet. 2009, 52, 77–87. [Google Scholar] [CrossRef]
- Hempel, M.; Rivera Brugués, N.; Wagenstaller, J.; Lederer, G.; Weitensteiner, A.; Seidel, H.; Meitinger, T.; Strom, T.M. Microdeletion Syndrome 16p11.2-P12.2: Clinical and Molecular Characterization. Am. J. Med. Genet. A 2009, 149A, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Shinawi, M.; Liu, P.; Kang, S.-H.L.; Shen, J.; Belmont, J.W.; Scott, D.A.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent Reciprocal 16p11.2 Rearrangements Associated with Global Developmental Delay, Behavioural Problems, Dysmorphism, Epilepsy, and Abnormal Head Size. J. Med. Genet. 2010, 47, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, S.; Reymond, A.; Zufferey, F.; Harewood, L.; Walters, R.G.; Kutalik, Z.; Martinet, D.; Shen, Y.; Valsesia, A.; Beckmann, N.D.; et al. Mirror Extreme BMI Phenotypes Associated with Gene Dosage at the Chromosome 16p11.2 Locus. Nature 2011, 478, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, R.G.; Jacquemont, S.; Valsesia, A.; de Smith, A.J.; Martinet, D.; Andersson, J.; Falchi, M.; Chen, F.; Andrieux, J.; Lobbens, S.; et al. A New Highly Penetrant Form of Obesity Due to Deletions on Chromosome 16p11.2. Nature 2010, 463, 671–675. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, S.E.; Makarov, V.; Kirov, G.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; Krastoshevsky, O.; et al. Microduplications of 16p11.2 Are Associated with Schizophrenia. Nat. Genet. 2009, 41, 1223–1227. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A Recurrent 16p12.1 Microdeletion Supports a Two-Hit Model for Severe Developmental Delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef]
- Andersen, E.F.; Baldwin, E.E.; Ellingwood, S.; Smith, R.; Lamb, A.N. Xq28 Duplication Overlapping the Int22h-1/Int22h-2 Region and Including RAB39B and CLIC2 in a Family with Intellectual and Developmental Disability. Am. J. Med. Genet. A 2014, 164A, 1795–1801. [Google Scholar] [CrossRef]
- Piton, A.; Redin, C.; Mandel, J.-L. XLID-Causing Mutations and Associated Genes Challenged in Light of Data from Large-Scale Human Exome Sequencing. Am. J. Hum. Genet. 2013, 93, 368–383. [Google Scholar] [CrossRef] [Green Version]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2012, 2, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 Duplication Syndrome. Am. J. Med. Genet. A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Exposito-Alonso, D.; Rico, B. Mechanisms Underlying Circuit Dysfunction in Neurodevelopmental Disorders. Annu. Rev. Genet. 2022, 56, 391–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, Y.; Zhang, F.; Chiang, C.; Pillalamarri, V.; Blumenthal, I.; Talkowski, M.; Wu, B.-L.; Gusella, J.F. Molecular Analysis of a Deletion Hotspot in the NRXN1 Region Reveals the Involvement of Short Inverted Repeats in Deletion CNVs. Am. J. Hum. Genet. 2013, 92, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-Linked Genes Encoding Neuroligins NLGN3 and NLGN4 Are Associated with Autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.; Feng, G. SHANK Proteins: Roles at the Synapse and in Autism Spectrum Disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Motazacker, M.M.; Rost, B.R.; Hucho, T.; Garshasbi, M.; Kahrizi, K.; Ullmann, R.; Abedini, S.S.; Nieh, S.E.; Amini, S.H.; Goswami, C.; et al. A Defect in the Ionotropic Glutamate Receptor 6 Gene (GRIK2) Is Associated with Autosomal Recessive Mental Retardation. Am. J. Hum. Genet. 2007, 81, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA Receptor GluA2 Subunit Defects Are a Cause of Neurodevelopmental Disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [Green Version]
- Srour, M.; AlHakeem, A.; Shevell, M. Chapter 19—Global Developmental Delay and Intellectual Disability. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 269–281. ISBN 978-0-12-813955-4. [Google Scholar]
- Komiyama, N.H.; Watabe, A.M.; Carlisle, H.J.; Porter, K.; Charlesworth, P.; Monti, J.; Strathdee, D.J.C.; O’Carroll, C.M.; Martin, S.J.; Morris, R.G.M.; et al. SynGAP Regulates ERK/MAPK Signaling, Synaptic Plasticity, and Learning in the Complex with Postsynaptic Density 95 and NMDA Receptor. J. Neurosci. 2002, 22, 9721–9732. [Google Scholar] [CrossRef]
- Tomoda, T.; Kim, J.H.; Zhan, C.; Hatten, M.E. Role of Unc51.1 and Its Binding Partners in CNS Axon Outgrowth. Genes. Dev. 2004, 18, 541–558. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, F.F.; Daoud, H.; Piton, A.; Gauthier, J.; Dobrzeniecka, S.; Krebs, M.-O.; Joober, R.; Lacaille, J.-C.; Nadeau, A.; Milunsky, J.M.; et al. De Novo SYNGAP1 Mutations in Nonsyndromic Intellectual Disability and Autism. Biol. Psychiatry 2011, 69, 898–901. [Google Scholar] [CrossRef]
- Yoshida, T.; Yasumura, M.; Uemura, T.; Lee, S.-J.; Ra, M.; Taguchi, R.; Iwakura, Y.; Mishina, M. IL-1 Receptor Accessory Protein-like 1 Associated with Mental Retardation and Autism Mediates Synapse Formation by Trans-Synaptic Interaction with Protein Tyrosine Phosphatase δ. J. Neurosci. 2011, 31, 13485–13499. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A Systematic, Large-Scale Resequencing Screen of X-Chromosome Coding Exons in Mental Retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Verhage, M.; Sørensen, J.B. SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron 2020, 107, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Gauthier, J.; Dobrzeniecka, S.; Lortie, A.; Mottron, L.; Vanasse, M.; D’Anjou, G.; Lacaille, J.C.; Rouleau, G.A.; Michaud, J.L. Intellectual Disability without Epilepsy Associated with STXBP1 Disruption. Eur. J. Hum. Genet. 2011, 19, 607–609. [Google Scholar] [CrossRef]
- Merienne, K.; Jacquot, S.; Pannetier, S.; Zeniou, M.; Bankier, A.; Gecz, J.; Mandel, J.L.; Mulley, J.; Sassone-Corsi, P.; Hanauer, A. A Missense Mutation in RPS6KA3 (RSK2) Responsible for Non-Specific Mental Retardation. Nat. Genet. 1999, 22, 13–14. [Google Scholar] [CrossRef]
- Nakashima, M.; Kato, M.; Matsukura, M.; Kira, R.; Ngu, L.-H.; Lichtenbelt, K.D.; van Gassen, K.L.I.; Mitsuhashi, S.; Saitsu, H.; Matsumoto, N. De Novo Variants in CUL3 Are Associated with Global Developmental Delays with or without Infantile Spasms. J. Hum. Genet. 2020, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- da Silva Montenegro, E.M.; Costa, C.S.; Campos, G.; Scliar, M.; de Almeida, T.F.; Zachi, E.C.; Silva, I.M.W.; Chan, A.J.S.; Zarrei, M.; Lourenço, N.C.V.; et al. Meta-Analyses Support Previous and Novel Autism Candidate Genes: Outcomes of an Unexplored Brazilian Cohort. Autism. Res. 2020, 13, 199–206. [Google Scholar] [CrossRef]
- Hörnberg, H.; Pérez-Garci, E.; Schreiner, D.; Hatstatt-Burklé, L.; Magara, F.; Baudouin, S.; Matter, A.; Nacro, K.; Pecho-Vrieseling, E.; Scheiffele, P. Rescue of Oxytocin Response and Social Behaviour in a Mouse Model of Autism. Nature 2020, 584, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Yeung, W.L.; Ko, C.H.; Poon, P.M.; Tong, S.F.; Chan, K.Y.; Lo, I.F.; Chan, L.Y.; Hui, J.; Wong, V.; et al. Spectrum of Mutations in the MECP2 Gene in Patients with Infantile Autism and Rett Syndrome. J. Med. Genet. 2000, 37, E41. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare Coding Variation Provides Insight into the Genetic Architecture and Phenotypic Context of Autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Noor, A.; Degagne, B.; Baker, K.; Bok, L.A.; Brady, A.F.; Chitayat, D.; Chung, B.H.; Cytrynbaum, C.; Dyment, D.; et al. Phenotypic Spectrum Associated with PTCHD1 Deletions and Truncating Mutations Includes Intellectual Disability and Autism Spectrum Disorder. Clin. Genet. 2015, 88, 224–233. [Google Scholar] [CrossRef]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, R.T.; Chantranupong, L.; Hakim, R.; Levasseur, J.; Wang, W.; Merchant, T.; Gorman, K.; Budnik, B.; Sabatini, B.L. Abnormal Striatal Development Underlies the Early Onset of Behavioral Deficits in Shank3B−/− Mice. Cell Rep. 2019, 29, 2016–2027.e4. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, R.T.; Wang, W.; Croney, D.M.; Kozorovitskiy, Y.; Sabatini, B.L. Early Hyperactivity and Precocious Maturation of Corticostriatal Circuits in Shank3B−/− Mice. Nat. Neurosci. 2016, 19, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Bariselli, S.; Tzanoulinou, S.; Glangetas, C.; Prévost-Solié, C.; Pucci, L.; Viguié, J.; Bezzi, P.; O’Connor, E.C.; Georges, F.; Lüscher, C.; et al. SHANK3 Controls Maturation of Social Reward Circuits in the VTA. Nat. Neurosci. 2016, 19, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Kao, F.-C.; Su, S.-H.; Carlson, G.C.; Liao, W. MeCP2-Mediated Alterations of Striatal Features Accompany Psychomotor Deficits in a Mouse Model of Rett Syndrome. Brain Struct. Funct. 2015, 220, 419–434. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.; Stockler-Ipsiroglu, S. Early Identification of Treatable Inborn Errors of Metabolism in Children with Intellectual Disability: The Treatable Intellectual Disability Endeavor Protocol in British Columbia. Paediatr. Child Health 2014, 19, 469–471. [Google Scholar] [CrossRef] [Green Version]
- Silove, N.; Collins, F.; Ellaway, C. Update on the Investigation of Children with Delayed Development. J. Paediatr. Child Health 2013, 49, 519–525. [Google Scholar] [CrossRef]
- Tirosh, E.; Jaffe, M. Global Developmental Delay and Mental Retardation—A Pediatric Perspective. Dev. Disabil. Res. Rev. 2011, 17, 85–92. [Google Scholar] [CrossRef]
- Mithyantha, R.; Kneen, R.; McCann, E.; Gladstone, M. Current Evidence-Based Recommendations on Investigating Children with Global Developmental Delay. Arch. Dis. Child 2017, 102, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbruggen, K.T.; Meiners, L.C.; Sijens, P.E.; Lunsing, R.J.; van Spronsen, F.J.; Brouwer, O.F. Magnetic Resonance Imaging and Proton Magnetic Resonance Spectroscopy of the Brain in the Diagnostic Evaluation of Developmental Delay. Eur. J. Paediatr. Neurol. 2009, 13, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Batty, R.; Warren, D.; Hart, A.; Sharrard, M.; Mordekar, S.R.; Raghavan, A.; Connolly, D.J.A. The Use of MR Imaging and Spectroscopy of the Brain in Children Investigated for Developmental Delay: What Is the Most Appropriate Imaging Strategy? Eur. Radiol. 2011, 21, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.I.; Majnemer, A.; Rosenbaum, P.; Abrahamowicz, M. Etiologic Yield of Subspecialists’ Evaluation of Young Children with Global Developmental Delay. J. Pediatr. 2000, 136, 593–598. [Google Scholar] [CrossRef]
- Shevell, M. The Role of the Pediatric Neurologist in the Care of Children with Neurodevelopmental Disabilities. Pediatr. Neurol. 2018, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kostanjsek, N. Use of The International Classification of Functioning, Disability and Health (ICF) as a Conceptual Framework and Common Language for Disability Statistics and Health Information Systems. BMC Public Health 2011, 11 (Suppl. 4), S3. [Google Scholar] [CrossRef] [Green Version]
- Fehlings, D.L.; Penner, M.; Donner, E.J.; Shevell, M.I. Management of Common Comorbidities Associated with Neurodevelopmental Disorders. In Swaiman’s Pediatric Neurology, 6th ed.; Swaiman, K.F., Ashwal, S., Ferriero, D.M., Schor, N.F., Finkel, R.S., Gropman, A.L., Pearl, P.L., Shevell, M.I., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 472–477. ISBN 978-0-323-37101-8. [Google Scholar]
- Bruni, O.; Angriman, M.; Melegari, M.G.; Ferri, R. Pharmacotherapeutic Management of Sleep Disorders in Children with Neurodevelopmental Disorders. Expert Opin. Pharmacother. 2019, 20, 2257–2271. [Google Scholar] [CrossRef]

| Common Pathways | Genes | Function |
|---|---|---|
| Synaptic Signaling | NRX1 | Cell-surface receptors that bind neuroligins; required for efficient neurotransmission. |
| NLGN3 NLGLN4 | Mediate cell-to-cell interactions between neurons; linked to glutamatergic postsynaptic proteins. | |
| SHANK2 SHANK3 | Scaffolding and cell adhesion proteins; required for synaptic plasticity. | |
| GRI2K GRIN2B GRIA2 | Subunits of synaptic glutamate receptors; required for neurotransmission. | |
| SYNGAP1 | Part of the NMDA receptor complex; involved in negative regulation of ERK/MAPK pathway. | |
| IL1RAPL1 | Part of the interleukin 1 receptor; required for neuronal calcium-regulated vesicle release and dendrite differentiation. | |
| CASK | Part of the MAGUK family; scaffolding proteins. | |
| STXBP1 | Synaptic vesicle docking and fusion; required for efficient neurotransmission. | |
| Protein Homeostasis | RPS6KA3 | Part of the RSK (ribosomal S6 kinase) family of growth-factor-regulated serine/threonine kinases; involved in ERK/MAPK pathway. |
| CUL3 | Part of the ubiquitin-proteasome system; required for proteasomal degradation of unwanted proteins. | |
| Epigenetic Regulation | MECP2 | Chromatin-associated protein involved in methyl binding to control transcription; required for maturation of neurons. |
| CHD8 | ATP-dependent chromatin-remodeling factor that regulates transcription. |
| Evaluation | Recommendation |
|---|---|
| 1. Detailed history including developmental and family history with thorough clinical examination |
|
| 2. First tier genetic testing |
|
| 3. Second tier genetic testing |
|
| 4. Further investigations |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlMutiri, R.; Malta, M.; Shevell, M.I.; Srour, M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children 2023, 10, 414. https://doi.org/10.3390/children10030414
AlMutiri R, Malta M, Shevell MI, Srour M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children. 2023; 10(3):414. https://doi.org/10.3390/children10030414
Chicago/Turabian StyleAlMutiri, Rowim, Maisa Malta, Michael I. Shevell, and Myriam Srour. 2023. "Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability" Children 10, no. 3: 414. https://doi.org/10.3390/children10030414
APA StyleAlMutiri, R., Malta, M., Shevell, M. I., & Srour, M. (2023). Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children, 10(3), 414. https://doi.org/10.3390/children10030414