
The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Comorbidities
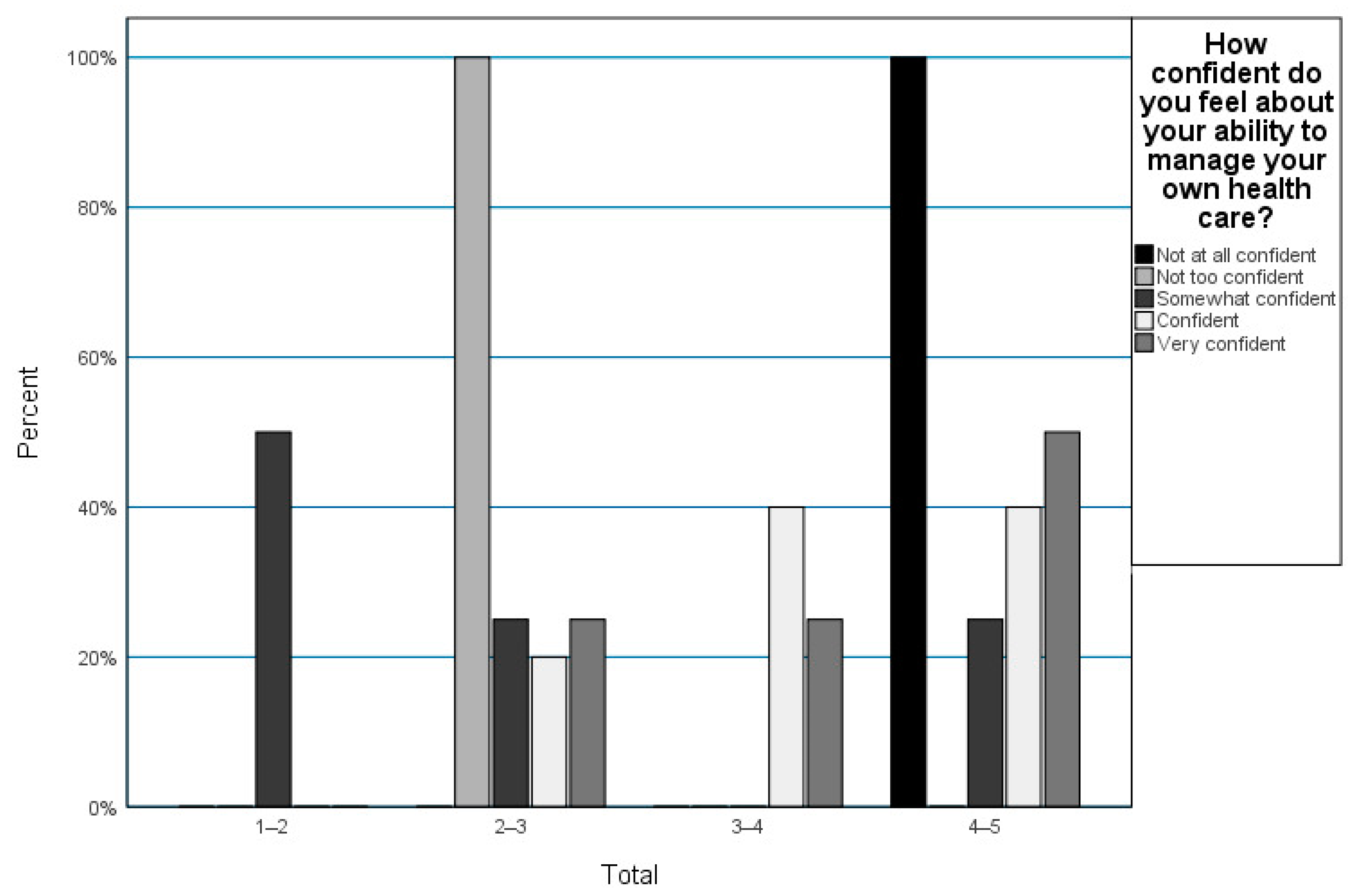
3.2. Transition Readiness
4. Discussion
4.1. Cardiac Management
4.2. Respiratory Management
4.3. Rehabilitation and Orthopedic Management
4.4. Endocrinological Management
4.5. Gastro-Intestinal and Renal Management
4.6. Psychological and Psychiatric Management
4.7. Socio-Economic Aspects
4.8. Palliative Care
4.9. Study Limitations
4.10. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wasilewska, E.; Małgorzewicz, S.; Sobierajska-Rek, A.; Jabłońska-Brudło, J.; Górska, L.; Śledzińska, K.; Bautembach-Minkowska, J.; Wierzba, J. Transition from Childhood to Adulthood in Patients with Duchenne Muscular Dystrophy. Medicina 2020, 56, 426. [Google Scholar] [CrossRef]
- LaPelusa, A.; Kentris, M. Muscular Dystrophy. [Updated 2023 Feb 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2. Available online: https://www.ncbi.nlm.nih (accessed on 8 March 2023).
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The Burden, Epidemiology, Costs and Treatment for Duchenne Muscular Dystrophy: An Evidence Review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Epure, D.A.-M.; Vladâcenco, O.A.; Lupu, M. Esențialul În Neurologia Pediatrică, 1st ed.; Editura Universitară “Carol Davila” Bucureşti a Universităţii de Medicină şi Farmacie “Carol Davila”: Bucharest, Romania, 2022; Volume 1. [Google Scholar]
- Broomfield, J.; Hill, M.; Guglieri, M.; Crowther, M.; Abrams, K. Life Expectancy in Duchenne Muscular Dystrophy. Neurology 2021, 97, E2304–E2314. [Google Scholar] [CrossRef] [PubMed]
- Klimchak, A.C.; Shelagh, M.; Szabo, M.; Qian, C.; Popoff, E.; Iannaccone, S.; Gooch, K.L. Characterizing Demographics, Comorbidities, and Costs of Care among Populations with Duchenne Muscular Dystrophy with Medicaid and Commercial Coverage. J. Manag. Care Spec. Pharm. 2021, 27, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kawai, M.; Kimura, E.; Ogata, K.; Takahashi, T.; Kobayashi, M.; Takada, H.; Kuru, S.; Mikata, T.; Matsumura, T.; et al. Study of Duchenne Muscular Dystrophy Long-Term Survivors Aged 40 Years and Older Living in Specialized Institutions in Japan. Neuromuscul. Disord. 2017, 27, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, Y.; Mistry, B.; Gibson, B.E. Transitioning to Adulthood with a Progressive Condition: Best Practice Assumptions and Individual Experiences of Young Men with Duchenne Muscular Dystrophy. Disabil. Rehabil. 2015, 37, 1144–1151. [Google Scholar] [CrossRef]
- Rosen, D.S.; Blum, R.W.; Britto, M.; Sawyer, S.M.; Siegel, D.M. Transition to Adult Health Care for Adolescents and Young Adults with Chronic Conditions: Position Paper of the Society for Adolescent Medicine. J. Adolesc. Health 2003, 33, 309–311. [Google Scholar] [CrossRef]
- Rodger, S.; Steffensen, B.F.; Lochmüller, H. Transition from Childhood to Adulthood in Duchenne Muscular Dystrophy (DMD). Orphanet J. Rare Dis. 2012, 7, A8. [Google Scholar] [CrossRef]
- White, P.; Schmidt, A.; Ilango, S.; Shorr, J.; Beck, D.; McManus, M. Six Core Elements of Health Care Transition™ 3.0: An Implementation Guide. Washington, DC: Got Transition, The National Alliance to Advance Adolescent Health. July 2020. Available online: www.IHI.org (accessed on 9 March 2023).
- Abbott, D. Other Voices, Other Rooms: Reflections on Talking to Young Men with Duchenne Muscular Dystrophy and Their Families About Transition to Adulthood. Child. Soc. 2012, 26, 241–250. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Respiratory, Cardiac, Bone Health, and Orthopaedic Management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Rodger, S.; Woods, K.L.; Bladen, C.L.; Stringer, A.; Vry, J.; Gramsch, K.; Kirschner, J.; Thompson, R.; Bushby, K.; Lochmüller, H. Adult Care for Duchenne Muscular Dystrophy in the UK. J. Neurol. 2015, 262, 629–641. [Google Scholar] [CrossRef]
- Tapawan, S.J.C.; Wang, F.S.; Lee, M.W.; Chua, A.Q.; Lin, J.B.; Han, V.; Lim, M.T.; Ong, H.T.; Tay, S.K. Perspectives on Palliative Care Among Duchenne Muscular Dystrophy Patients and Their Families in Singapore. Ann. Acad. Med. Singapore. 2020, 49, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.L.; Sawicki, G.S.; Miller, M.D.; Smotherman, C.; Lukens-Bull, K.; Livingood, W.C.; Ferris, M.; Kraemer, D.F. The Transition Readiness Assessment Questionnaire (TRAQ): Its Factor Structure, Reliability, and Validity. Acad. Pediatr. 2014, 14, 415–422. [Google Scholar] [CrossRef]
- Adam, M.P.; Everman, D.B.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. (Eds.) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. PMID: 20301490. Available online: https://www.genome.jp/dbget-bin/www_bget?refseq:NP_001116538 (accessed on 9 March 2023).
- Lee, I.; Turnage, C.; Sutyla, R.; Mitchell, P.; Lindahl, H.; Jesus, A.; Scharf, R.J. The Hidden Disease: Delayed Diagnosis in Duchenne Muscular Dystrophy and Co-Occurring Conditions. J. Dev. Behav. Pediatr. 2022, 43, e541–e545. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, R.M. Age at Diagnosis for Duchenne Muscular Dystrophy: Why We Must Do Better. Muscle Nerve 2022, 66, 116–117. [Google Scholar] [CrossRef]
- Thomas, S.; Conway, K.M.; Fapo, O.; Street, N.; Mathews, K.D.; Mann, J.R.; Romitti, P.A.; Soim, A.; Westfield, C.; Fox, D.J.; et al. Time to Diagnosis of Duchenne Muscular Dystrophy Remains Unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015. Muscle Nerve 2022, 66, 193–197. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 3: Primary Care, Emergency Management, Psychosocial Care, and Transitions of Care across the Lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, M.; Coskun, B.; Stolte, B.; Della-Marina, A.; Kölbel, H.; Lax, H.; Nonnemacher, M.; Kleinschnitz, C.; Schara-Schmidt, U.; Hagenacker, T. Essen Transition Model for Neuromuscular Diseases. Neurol. Res. Pract. 2022, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Verhaert, D.; Richards, K.; Rafael-Fortney, J.A.; Raman, S.V. Cardiac Involvement in Patients with Muscular Dystrophies. Circ. Cardiovasc. Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Implementation of Multidisciplinary Care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Ararat, E.; Mhanna, M.J. Cardiac Phenotype Determines Survival in Duchenne Muscular Dystrophy. Pediatr. Pulmonol. 2016, 51, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Wittlieb-Weber, C.A.; Knecht, K.R.; Villa, C.R.; Cunningham, C.; Conway, J.; Bock, M.J.; Gambetta, K.E.; Lal, A.K.; Schumacher, K.R.; Law, S.P.; et al. Risk Factors for Cardiac and Non-Cardiac Causes of Death in Males with Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2020, 41, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, D.; Khan, S.; Khera, R.; Bhatt, A.; Garg, S.; Grodin, J.L.; Morlend, R.; Araj, F.G.; Amin, A.A.; Thibodeau, J.T.; et al. Predictors of Death in Adults with Duchenne Muscular Dystrophy–Associated Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e006340. [Google Scholar] [CrossRef] [PubMed]
- Aliverti, A.; LoMauro, A.; D’Angelo, M.G. Assessment and Management of Respiratory Function in Patients with Duchenne Muscular Dystrophy: Current and Emerging Options. Ther. Clin. Risk Manag. 2015, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O.H.; Finkel, R.S.; Rummey, C.; Benton, M.J.; Glanzman, A.M.; Flickinger, J.; Lindström, B.-M.; Meier, T. Characterization of Pulmonary Function in Duchenne Muscular Dystrophy. Pediatr. Pulmonol. 2015, 50, 487–494. [Google Scholar] [CrossRef]
- Chindriş, S.; Daviţoiu, A.M.; Spătariu, L.; Buzoianu, E.; Iancu, M.; Ţincu, I.; Ghiorghiu, I.A.; Zamfirescu, A.; Vlad, G.; Albert, M.; et al. A Review of Respiratory Management in Children with Duchenne Muscular Dystrophy. Pediatruro 2021, 1, 18. [Google Scholar] [CrossRef]
- Case, L.E.; Apkon, S.D.; Eagle, M.; Gulyas, A.; Juel, L.; Matthews, D.; Newton, R.A.; Posselt, H.F. Rehabilitation Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. S2), S17–S33. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Hadjiyannakis, S.; McMillan, H.J.; Noritz, G.; Weber, D.R. Bone Health and Osteoporosis Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. S2), S34–S42. [Google Scholar] [CrossRef]
- Connolly, A.M.; Malkus, E.C.; Mendell, J.R.; Flanigan, K.M.; Miller, J.P.; Schierbecker, J.R.; Siener, C.A.; Golumbek, P.T.; Zaidman, C.M.; Mcdonald, C.M.; et al. Outcome Reliability in Non-Ambulatory Boys/Men with Duchenne Muscular Dystrophy. Muscle Nerve 2015, 51, 522–532. [Google Scholar] [CrossRef]
- Goemans, N. How Glucocorticoids Change Life in Duchenne Muscular Dystrophy. Lancet 2018, 391, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Cruz Guzmán, O. del R.; Chávez García, A.L.; Rodríguez-Cruz, M. Muscular Dystrophies at Different Ages: Metabolic and Endocrine Alterations. Int. J. Endocrinol. 2012, 2012, 485376. [Google Scholar] [CrossRef]
- Bianchi, M.L.; Biggar, D.; Bushby, K.; Rogol, A.D.; Rutter, M.M.; Tseng, B. Endocrine Aspects of Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2011, 21, 298–303. [Google Scholar] [CrossRef]
- Weber, D.R.; Hadjiyannakis, S.; McMillan, H.J.; Noritz, G.; Ward, L.M. Obesity and Endocrine Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. 2), S43–S52. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, D.; Watne, L.; Gottrand, F.; Gulyas, A.; Kaul, A.; Larson, J.; Tomezsko, J. Nutritional and Gastrointestinal Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. S2), S53–S61. [Google Scholar] [CrossRef]
- Lo Cascio, C.M.; Goetze, O.; Latshang, T.D.; Bluemel, S.; Frauenfelder, T.; Bloch, K.E. Gastrointestinal Dysfunction in Patients with Duchenne Muscular Dystrophy. PLoS ONE 2016, 11, e0163779. [Google Scholar] [CrossRef]
- Kutluk, M.G.; Doğan, Ç.S. Kidney Involvement and Associated Risk Factors in Children with Duchenne Muscular Dystrophy. Pediatr. Nephrol. 2020, 35, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N. Combining Genetics, Neuropsychology and Neuroimaging to Improve Understanding of Brain Involvement in Duchenne Muscular Dystrophy—A Narrative Review. Neuromuscul. Disord. 2020, 30, 437–442. [Google Scholar] [CrossRef]
- Doorenweerd, N.; Straathof, C.S.; Dumas, E.M.; Spitali, P.; Ginjaar, I.B.; Wokke, B.H.; Schrans, D.G.; van den Bergen, J.C.; van Zwet, E.W.; Webb, A.; et al. Reduced Cerebral Gray Matter and Altered White Matter in Boys with Duchenne Muscular Dystrophy. Ann. Neurol. 2014, 76, 403–411. [Google Scholar] [CrossRef]
- Di Filippo, T.; Parisi, L.; Roccella, M. Psychological Aspects in Children Affected by Duchenne de Boulogne Muscular Dystrophy. Ment. Illn. 2012, 4, 21–24. [Google Scholar] [CrossRef]
- Colvin, M.K.; Poysky, J.; Kinnett, K.; Damiani, M.; Gibbons, M.; Hoskin, J.; Moreland, S.; Trout, C.J.; Weidner, N. Psychosocial Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. S2), S99–S109. Available online: http://publications.aap.org/pediatrics/article-pdf/142/Supplement_2/S99/904208/peds_20180333l.pdf (accessed on 7 March 2023). [CrossRef] [PubMed]
- Fujino, H.; Iwata, Y.; Saito, T.; Matsumura, T.; Fujimura, H.; Imura, O. The Experiences of Patients with Duchenne Muscular Dystrophy in Facing and Learning about Their Clinical Conditions. Int. J. Qual. Stud. Health Well-Being 2016, 11, 32045. [Google Scholar] [CrossRef]
- Donaldson, A.; Guntrum, D.; Ciafaloni, E.; Statland, J. Achieving Life Milestones in Duchenne/Becker Muscular Dystrophy. Neurol. Clin. Pract. 2021, 11, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Trout, C.J.; Case, L.E.; Clemens, P.R.; McArthur, A.; Noritz, G.; Ritzo, M.; Wagner, K.R.; Vroom, E.; Kennedy, A. A Transition Toolkit for Duchenne Muscular Dystrophy. Pediatrics 2018, 142 (Suppl. 2), S110–S117. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Suzuki, M. Independent Living with Duchenne Muscular Dystrophy and Home Mechanical Ventilation in Areas of Japan with Insufficient National Welfare Services. Int. J. Qual. Stud. Health Well-Being 2013, 8, 20914. [Google Scholar] [CrossRef]
- Abbott, D.; Carpenter, J.; Bushby, K. Transition to Adulthood for Young Men with Duchenne Muscular Dystrophy: Research from the UK. Neuromuscul. Disord. 2012, 22, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Schrans, D.G.M.; Abbott, D.; Peay, H.L.; Pangalila, R.F.; Vroom, E.; Goemans, N.; Vles, J.S.H.; Aldenkamp, A.P.; Hendriksen, J.G.M. Transition in Duchenne Muscular Dystrophy: An Expert Meeting Report and Description of Transition Needs in an Emergent Patient Population: (Parent Project Muscular Dystrophy Transition Expert Meeting 17–18 June 2011, Amsterdam, The Netherlands). Neuromuscul. Disord. 2013, 23, 283–286. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; American Academy of Family Physicians; American College of Physicians-American Society of Internal Medicine. A Consensus Statement on Health Care Transitions for Young Adults with Special Health Care Needs. Pediatrics 2002, 110 Pt 2, 1304–1306. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Summary Statistics | |
|---|---|
| Age (median, range) | 17 (14–22) |
| Age at diagnosis (median, range) | 6 (1–8) |
| Living location (n, %) | |
| Urban | 6 (40) |
| Rural | 9 (60) |
| Comorbidities (n, %) | |
| Cardiac | 8 (53.3) |
| Pulmonary | 13 (86.7) |
| Orthopedic | 15 (100) |
| Scoliosis | 15 (100) |
| Club foot | 6 (40) |
| Cushing’s syndrome | 4 (26.7) |
| Obesity | 7 (46.7) |
| Emotional disorder | 10 (66.7) |
| Age at onset of comorbidities (median, range) | |
| Cardiac | 13.5 (11–15) |
| Pulmonary | 13 (10–17) |
| Mobility (n, %) | |
| Non-ambulatory | 6 (40%) |
| Ambulatory | 9 (60%) |
| Age non-ambulatory (mean, min-max) | 13.5 (10–17) |
| Mean | Median | Maximum | Minimum | Range | |
|---|---|---|---|---|---|
| Managing Medications | 2.9 | 2.6 | 4.8 | 1.0 | 3.8 |
| Appointment Keeping | 3.4 | 3.5 | 5.0 | 1.0 | 4.0 |
| Tracking Health Issues | 3.2 | 3.1 | 5.0 | 1.1 | 3.9 |
| Talking with Providers | 4.0 | 4.0 | 5.0 | 1.2 | 3.8 |
| Total TRAQ score | 3.3 | 3.6 | 4.8 | 1.3 | 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lupu, M.; Ioghen, M.; Perjoc, R.-Ș.; Scarlat, A.-M.; Vladâcenco, O.A.; Roza, E.; Epure, D.A.-M.; Teleanu, R.I.; Severin, E.M. The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases. Children 2023, 10, 959. https://doi.org/10.3390/children10060959
Lupu M, Ioghen M, Perjoc R-Ș, Scarlat A-M, Vladâcenco OA, Roza E, Epure DA-M, Teleanu RI, Severin EM. The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases. Children. 2023; 10(6):959. https://doi.org/10.3390/children10060959
Chicago/Turabian StyleLupu, Maria, Mihaela Ioghen, Radu-Ștefan Perjoc, Andra-Maria Scarlat, Oana Aurelia Vladâcenco, Eugenia Roza, Diana Ana-Maria Epure, Raluca Ioana Teleanu, and Emilia Maria Severin. 2023. "The Importance of Implementing a Transition Strategy for Patients with Muscular Dystrophy: From Child to Adult—Insights from a Tertiary Centre for Rare Neurological Diseases" Children 10, no. 6: 959. https://doi.org/10.3390/children10060959