


A Study of Maternal Patients Diagnosed with Inborn Errors of Metabolism Due to Positive Newborn Mass Screening in Their Newborns
 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
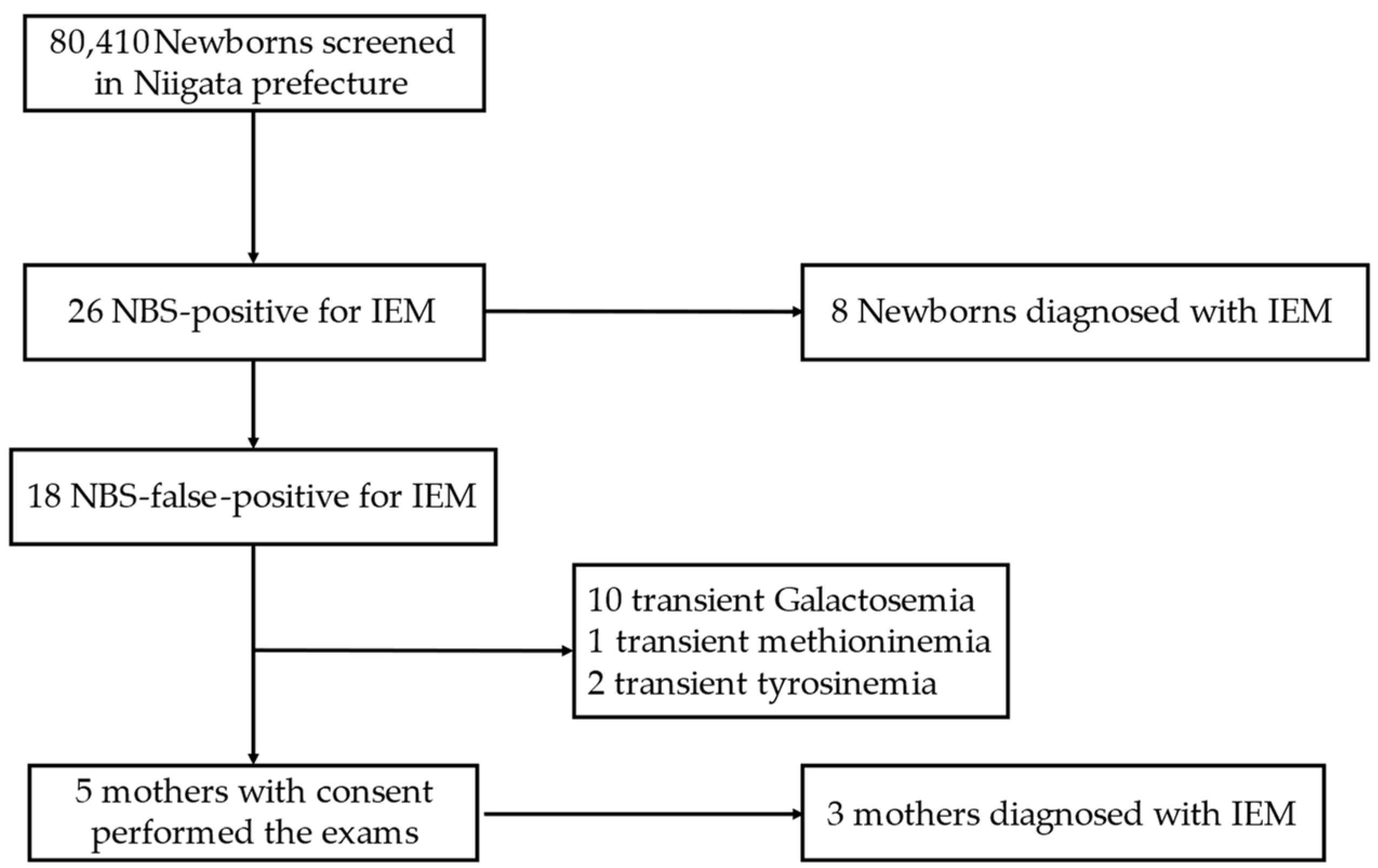
2.1. NBS in Niigata Prefecture
2.2. Subjects of This Study
3. Results
3.1. Patient 1
3.2. Patient 2
3.3. Patient 3
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lund, A.M.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Dunø, M.; Skogstrand, K.; Olsen, R.K.; Jensen, U.G.; Cohen, A.; et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland—Experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, K.; Zheng, Z.; Chen, Y.; Fu, C.; Lin, Y.; Chen, D. Newborn screening for primary carnitine deficiency in Quanzhou. China Clin. Chim. Acta 2021, 512, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Li, H.; Huang, T.; Zhang, Y.; Wang, C.; Gu, M. Biochemical, Molecular, and Clinical Characterization of Patients With Primary Carnitine Deficiency via Large-Scale Newborn Screening in Xuzhou Area. Front. Pediatr. 2019, 26, 50. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T. Newborn Screening in Japan-2021. Int. J. Neonatal Screen. 2022, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Tang, N.L.; Chien, Y.H.; Chen, C.A.; Lin, S.J.; Chiu, P.C.; Huang, A.C.; Hwu, W.L. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol. Genet. Metab. 2010, 100, 46–50. [Google Scholar] [CrossRef] [PubMed]
- De Biase, I.; Champaigne, N.L.; Schroer, R.; Pollard, L.M.; Longo, N.; Wood, T. Primary Carnitine Deficiency Presents Atypically with Long QT Syndrome: A Case Report. JIMD Rep. 2012, 2, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crombez, E.A.; Cederbaum, S.D.; Spector, E.; Chan, E.; Salazar, D.; Neidich, J.; Goodman, S. Maternal glutaric acidemia, type I identified by newborn screening. Mol. Genet. Metab. 2008, 94, 132–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyhan, W.L.; Willis, M.; Barshop, B.A.; Gangoiti, J. Positive newborn screen in the biochemically normal infant of a mother with treated holocarboxylase synthetase deficiency. J. Inherit. Metab. Dis. 2009, 32, S79–S82. [Google Scholar] [CrossRef] [PubMed]
- Leydiker, K.B.; Neidich, J.A.; Lorey, F.; Barr, E.M.; Puckett, R.L.; Lobo, R.M.; Abdenur, J.E. Maternal medium-chain acyl-CoA dehydrogenase deficiency identified by newborn screening. Mol. Genet. Metab. 2011, 103, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Aksglaede, L.; Christensen, M.; Olesen, J.H.; Duno, M.; Olsen, R.K.; Andresen, B.S.; Hougaard, D.M.; Lund, A.M. Abnormal Newborn Screening in a Healthy Infant of a Mother with Undiagnosed Medium-Chain Acyl-CoA Dehydrogenase Deficiency. JIMD Rep. 2015, 23, 67–70. [Google Scholar] [CrossRef] [PubMed]
- McGoey, R.R.; Marble, M. Positive newborn screen in a normal infant of a mother with asymptomatic very long-chain Acyl-CoA dehydrogenase deficiency. J. Pediatr. 2011, 158, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Neidich, J.A.; Salazar, D.; Thomas-Johnson, E.; Ferreira, B.F.; Kwong, A.M.; Lin, A.M.; Jonas, A.J.; Levine, S.; Lorey, F.; et al. Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (cblC) detected through low carnitine levels on newborn screening. J. Pediatr. 2009, 155, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.; Yonezawa, A.; Masuda, S.; Inui, K.; Sim, K.G.; Carpenter, K.; Olsen, R.K.; Mitchell, J.J.; Rhead, W.J.; Peters, G.; et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2011, 32, E1976–E1984. [Google Scholar] [CrossRef] [PubMed]
- Frigeni, M.; Balakrishnan, B.; Yin, X.; Calderon, F.; Mao, R.; Pasquali, M.; Longo, N. Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 2017, 38, 1684–1699. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Chien, Y.H.; Chen, P.W.; Leung-Sang Tang, N.; Chiu, P.C.; Hwu, W.L.; Lee, N.C. Carnitine uptake defect (primary carnitine deficiency): Risk in genotype-phenotype correlation. Hum. Mutat. 2013, 34, 655. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Lund, A.M.; Risom, L.; Wibrand, F.; Gislason, H.; Nielsen, O.W.; Køber, L.; Duno, M. Residual OCTN2 transporter activity, carnitine levels and symptoms correlate in patients with primary carnitine deficiency. Mol. Genet. Metab. Rep. 2014, 22, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajcovicová-Kudlácková, M.; Simoncic, R.; Béderová, A.; Babinská, K.; Béder, I. Correlation of carnitine levels to methionine and lysine intake. Physiol. Res. 2000, 49, 399–402. [Google Scholar] [PubMed]


{kind=link}
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Diagnosis | PCD | PCD | 3-MCCD |
| Age(y) | 30 | 36 | 42 |
| Opportunity of diagnosis | Low C0 of NBS | Low C0 of NBS | Low C0 and high C5-OH of NBS |
| Clinical presentation | No symptoms | No symptoms | No symptoms |
| Examination | Free carnitine 8.6 μmol/L in blood | Free carnitine 5.9 μmol/L in blood | Free carnitine 4.4 μmol/L in blood 3-Methylcrotonylglycine and 3-Hydroxyisovaleric acid Elevation in urine |
| Gene analysis | SLC22A5 c.1063T > C/ c.1266A > G | SLC22A5 c.865C > T/ c.1400C > G | Not implemented |
| Diagnosis of newborn | Secondary to low maternal plasma carnitine levels | Secondary to low maternal plasma carnitine levels | Secondary to low maternal plasma carnitine levels |
| Treatments | Oral carnitine | Oral carnitine | Oral carnitine |
| Symptoms after treatment | Improvement in headache and fatigue | No change | Improvement in headache and fatigue |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onuki, T.; Hiroshima, S.; Sawano, K.; Shibata, N.; Ogawa, Y.; Nagasaki, K.; Nyuzuki, H. A Study of Maternal Patients Diagnosed with Inborn Errors of Metabolism Due to Positive Newborn Mass Screening in Their Newborns. Children 2023, 10, 1341. https://doi.org/10.3390/children10081341
Onuki T, Hiroshima S, Sawano K, Shibata N, Ogawa Y, Nagasaki K, Nyuzuki H. A Study of Maternal Patients Diagnosed with Inborn Errors of Metabolism Due to Positive Newborn Mass Screening in Their Newborns. Children. 2023; 10(8):1341. https://doi.org/10.3390/children10081341
Chicago/Turabian StyleOnuki, Takanori, Shota Hiroshima, Kentaro Sawano, Nao Shibata, Yohei Ogawa, Keisuke Nagasaki, and Hiromi Nyuzuki. 2023. "A Study of Maternal Patients Diagnosed with Inborn Errors of Metabolism Due to Positive Newborn Mass Screening in Their Newborns" Children 10, no. 8: 1341. https://doi.org/10.3390/children10081341
APA StyleOnuki, T., Hiroshima, S., Sawano, K., Shibata, N., Ogawa, Y., Nagasaki, K., & Nyuzuki, H. (2023). A Study of Maternal Patients Diagnosed with Inborn Errors of Metabolism Due to Positive Newborn Mass Screening in Their Newborns. Children, 10(8), 1341. https://doi.org/10.3390/children10081341