
Continuous Spike–Waves during Slow Sleep Today: An Update



Abstract
:
1. Introduction
2. Materials and Methods
3. Results
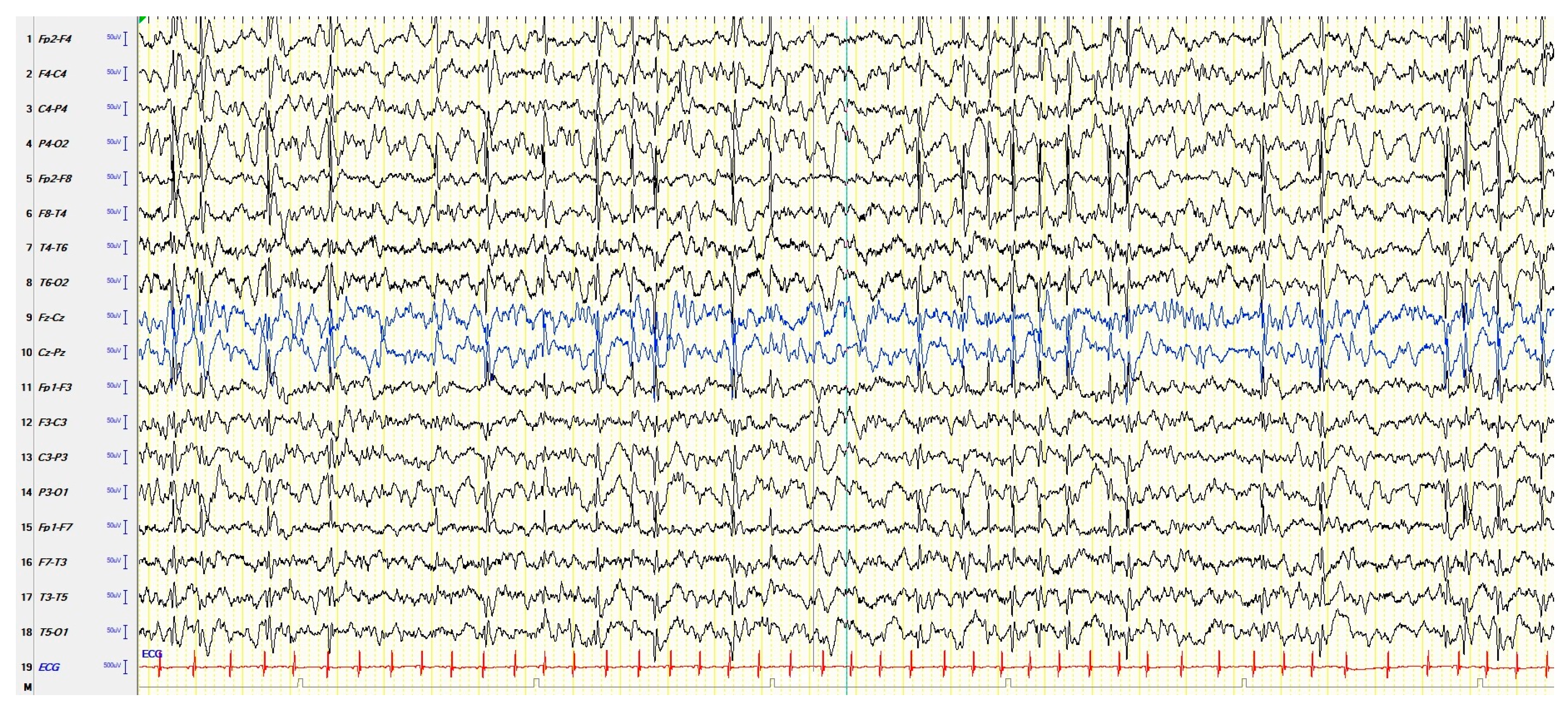
3.1. EEGs and Clinics of Typical and Atypical ESES
3.2. Predictive Factors of the Evolution into CSWS
3.3. Electrophysiology and Functional Neuroimaging
3.4. Treatments Regardless of Neurosurgery
3.5. Neurosurgical Treatment
3.6. Evolution and Prognostic Factors
3.7. Landau–Kleffner Syndrome (LKS)
3.7.1. Clinics, Epidemiology, and Differential Diagnosis
3.7.2. Neurophysiological Findings
3.7.3. Etiology
3.7.4. Physiopathology
3.7.5. Outcome
3.8. Treatments for LKS
3.9. Etiopathogenetic Factors of CSWS (Including ESES and LKS)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmitt, B. Sleep and epilepsy syndromes. Neuropediatrics 2015, 46, 171–180. [Google Scholar] [CrossRef]
- Shbarou, R.; Mikati, M.A. The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin. Pediatr. Neurol. 2016, 23, 134–142. [Google Scholar] [CrossRef]
- Besag, F.M. Epilepsy in patients with autism: Links, risks and treatment challenges. Neuropsychiatr. Dis. Treat. 2017, 14, 1–10. [Google Scholar] [CrossRef]
- Appendino, J.P.; Appendino, J.I. Encefalopatías epilépticas determinadas genéticamente [Genetically determined epileptic encephalopathies]. Medicina 2019, 79 (Suppl. 3), 42–47. [Google Scholar]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef]
- Van Bogaert, P. Long-term outcome of developmental and epileptic encephalopathies. Rev. Neurol. 2022, 178, 659–665. [Google Scholar] [CrossRef]
- Van Bogaert, P. Epilepsy syndromes of childhood with sleep activation: Insights from functional imaging. Eur. J. Paediatr. Neurol. 2020, 24, 58–60. [Google Scholar] [CrossRef]
- Auvin, S.; Cilio, M.R.; Vezzani, A. Current understanding and neurobiology of epileptic encephalopathies. Neurobiol. Dis. 2016, 92, 72–89. [Google Scholar] [CrossRef]
- Japaridze, N.; Menzel, E.; von Ondarza, G.; Steinmann, E.; Stephani, U. Risk factors of cognitive outcome in patients with atypical benign partial epilepsy/pseudo-Lennox syndrome (ABPE/PLS) and continues spike and wave during sleep (CSWS). Eur. J. Paediatr. Neurol. 2014, 18, 368–375. [Google Scholar] [CrossRef]
- Kennedy, A.; Hill, D. Dementia infantilis with cortical dysrhythmia. Arch. Dis. Child. 1942, 17, 122–129. [Google Scholar] [CrossRef]
- Patry, G.; Lyagoubi, S.; Tassinari, C.A. Subclinical “electrical status epilepticus” induced by sleep in children. A clinical and electroencephalographic study of six cases. Arch. Neurol. 1971, 24, 242–252. [Google Scholar] [CrossRef]
- Tassinari, C.A.; Rubboli, G.; Volpi, L.; Meletti, S.; d’Orsi, G.; Franca, M.; Sabetta, A.R.; Riguzzi, P.; Gardella, E.; Zaniboni, A.; et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin. Neurophysiol. 2000, 111 (Suppl. 2), S94–S102. [Google Scholar] [CrossRef]
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Azcona, G.; Gurtubay, I.G.; Mosquera, A.; Ibanez, B.; Cambra, K.; Aguilera-Albesa, S.; Yoldi-Petri, M.E. Estudio comparativo entre tres sistemas de cuantificacion del indice de punta-onda en pacientes con punta-onda continua del sueño lento [A comparative study of three systems for quantifying the spike and wave index in patients with continuous spikes and waves during slow sleep]. Rev. Neurol. 2017, 65, 439–446. [Google Scholar]
- Hirsch, E.; Caraballo, R.; Dalla Bernardina, B.; Loddenkemper, T.; Zuberi, S.M. Encephalopathy related to status epilepticus during slow sleep: From concepts to terminology. Epileptic Disord. 2019, 21, S5–S12. [Google Scholar] [CrossRef]
- Jansen, F.E.; Nikanorova, M.; Peltola, M. Current treatment options for encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, S76–S81. [Google Scholar] [CrossRef]
- Singhal, N.S.; Sullivan, J.E. Continuous spike-wave during slow wave sleep and related conditions. ISRN Neurol. 2014, 2014, 619079. [Google Scholar] [CrossRef]
- RamachandranNair, R. Encephalopathy associated with electrical status epilepticus of sleep (ESES): A practical approach. Indian J. Pediatr. 2020, 87, 1057–1061. [Google Scholar] [CrossRef]
- Sonnek, B.; Döring, J.H.; Mütze, U.; Schubert-Bast, S.; Bast, T.; Balke, D.; Reuner, G.; Schuler, E.; Klabunde-Cherwon, A.; Hoffmann, G.F.; et al. Clinical spectrum and treatment outcome of 95 children with continuous spikes and waves during sleep (CSWS). Eur. J. Paediatr. Neurol. 2021, 30, 121–127. [Google Scholar] [CrossRef]
- Rubboli, G.; Huber, R.; Tononi, G.; Tassinari, C.A. Encephalopathy related to status epilepticus during slow sleep: A link with sleep homeostasis? Epileptic Disord. 2019, 21, S62–S70. [Google Scholar] [CrossRef]
- Rubboli, G.; Gardella, E.; Cantalupo, G.; Tassinari, C.A. Encephalopathy related to status epilepticus during slow sleep (ESES). Pathophysiological insights and nosological considerations. Epilepsy Behav. 2023, 140, 109105. [Google Scholar] [CrossRef]
- Samanta, D.; Al Khalili, Y. Electrical status epilepticus in sleep. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nath, A.; Whitworth, E.; Bretz, D.; Davila-Williams, D.; McIntosh, L. Electrical status epilepticus in sleep (ESES) in an elderly adult: A case report. Cureus 2022, 14, e26372. [Google Scholar] [CrossRef]
- Rubboli, G.; Tassinari, C.A. Linking epilepsy, sleep disruption and cognitive impairment in encephalopathy related to status epilepticus during slow sleep (ESES). Epileptic Disord. 2019, 21, S1–S2. [Google Scholar] [CrossRef]
- Camfield, P.; Camfield, C. Regression in children with epilepsy. Neurosci. Biobehav. Rev. 2019, 96, 210–218. [Google Scholar] [CrossRef]
- Dorris, L.; O’Regan, M.; Wilson, M.; Zuberi, S.M. Progressive intellectual impairment in children with encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, S88–S96. [Google Scholar] [CrossRef]
- Jacob, J. Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia 2016, 57, 182–193. [Google Scholar] [CrossRef]
- Besag, F.; Aldenkamp, A.; Caplan, R.; Dunn, D.W.; Gobbi, G.; Sillanpää, M. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Epilepsy and autism. Epileptic Disord. 2016, 18 (Suppl. 1), S16–S23. [Google Scholar] [CrossRef]
- Besag, F.; Gobbi, G.; Aldenkamp, A.; Caplan, R.; Dunn, D.W.; Sillanpää, M. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Behavioural and psychiatric disorders associated with epilepsy syndromes. Epileptic Disord. 2016, 18 (Suppl. 1), S37–S48. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.; Liu, X.; Zhang, Y.; Jiang, Y.; Yang, Z. Epileptic negative myoclonus restricted to lower limbs in benign childhood focal epilepsy with vertex spikes. Eur. J. Neurol. 2019, 26, 1318–1326. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.P.; Zhang, Y.H.; Jiang, Y.W.; Yang, Z.X. Electroclinical characteristics of epilepsy children with midline epileptiform discharges related epileptic negative myoclonus as the first symptom. Chin. J. Pediatr. 2019, 57, 943–949. [Google Scholar]
- Aoun, M.A.; Eisermann, M.; Chemaly, N.; Losito, E.; Desguerre, I.; Nabbout, R.; Kaminska, A. Jerking during absences: Video-EEG and polygraphy of epileptic myoclonus associated with two paediatric epilepsy syndromes. Epileptic Disord. 2021, 23, 191–200. [Google Scholar] [CrossRef]
- Kalscheur, E.J.; Farias-Moeller, R.; Koop, J. Role of neuropsychology in identification of CSWS in a school-aged child with a remote neurological insult. Epilepsy Behav. Rep. 2021, 18, 100514. [Google Scholar] [CrossRef]
- Maltoni, L.; Posar, A.; Parmeggiani, A. Long-term follow-up of cognitive functions in patients with continuous spike-waves during sleep (CSWS). Epilepsy Behav. 2016, 60, 211–217. [Google Scholar] [CrossRef]
- Carvalho, D.; Mendonça, C.; Carvalho, J.; Martins, A.; Leal, A. High incidence of early thalamic lesions in the continuous spike-wave related with slow sleep (CSWS). Epilepsy Behav. 2023, 138, 109031. [Google Scholar] [CrossRef]
- Bennett-Back, O.; Uliel-Siboni, S.; Kramer, U. The yield of video-EEG telemetry evaluation for non-surgical candidate children. Eur. J. Paediatr. Neurol. 2016, 20, 848–854. [Google Scholar] [CrossRef]
- Nagyova, R.; Horsburgh, G.; Robertson, A.; Zuberi, S.M. The clinical utility of ambulatory EEG in childhood. Seizure 2019, 64, 45–49. [Google Scholar] [CrossRef]
- Tassinari, C.A.; Rubboli, G. Encephalopathy related to status epilepticus during slow sleep: Current concepts and future directions. Epileptic Disord. 2019, 21, S82–S87. [Google Scholar] [CrossRef]
- Belousova, E.D.; Ermakov, A. Épilepticheskaia éntsefalopatiia s prodolzhennoĭ spaĭk-vol-no-voĭ aktivnost’iu vo sne: Obzor literatury [Epileptic encephalopathy with continuous spikes and waves during sleep (CSWS): A review]. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova 2014, 114, 52–58. [Google Scholar]
- Altunel, A.; Altunel, E.Ö.; Sever, A. Response to adrenocorticotropic in attention deficit hyperactivity disorder-like symptoms in electrical status epilepticus in sleep syndrome is related to electroencephalographic improvement: A retrospective study. Epilepsy Behav. 2017, 74, 161–166. [Google Scholar] [CrossRef]
- van Iterson, L.; Vrij, S.; Sie, L.T.L.; Augustijn, P.B.; Rooze, A.C.S.; Jansen, F.E. Acquired visual agnosia as an uncommon presentation of epileptic encephalopathy in a 6-year-old boy with CSWS. Epilepsy Behav. Rep. 2021, 16, 100465. [Google Scholar] [CrossRef]
- Kuki, I.; Kawawaki, H.; Okazaki, S.; Ikeda, H.; Tomiwa, K. Epileptic encephalopathy with continuous spikes and waves in the occipito-temporal region during slow-wave sleep in two patients with acquired Kanji dysgraphia. Epileptic Disord. 2014, 16, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, C.A.; Cantalupo, G.; Rubboli, G. Focal ESES as a selective focal brain dysfunction: A challenge for clinicians, an opportunity for cognitive neuroscientists. Epileptic Disord. 2015, 17, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, E.; Møller, R.S.; Nikanorova, M.; Kölmel, M.S.; Stendevad, P.; Beniczky, S.; Tassinari, C.A.; Rubboli, G.; Gardella, E. Idiopathic encephalopathy related to status epilepticus during slow sleep (ESES) as a “pure” model of epileptic encephalopathy. An electroclinical, genetic, and follow-up study. Epilepsy Behav. 2019, 97, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Kowkabi, S.; Asadi-Pooya, A.A.; Malamiri, R.A.; Badv, R.S. Hemi-ESES associated with agenesis of the corpus callosum and normal cognition. Epilepsy Behav. Case Rep. 2019, 11, 96–98. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Veggiotti, P.; Kaltenmeier, M.C.; Piazza, E.; Gamboni, B.; Lopez Avaria, M.F.; Noli, D.; Adi, J.; Cersosimo, R. Encephalopathy with status epilepticus during sleep or continuous spikes and waves during slow sleep syndrome: A multicenter, long-term follow-up study of 117 patients. Epilepsy Res. 2013, 105, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Gencpinar, P.; Dundar, N.O.; Tekgul, H. Electrical status epilepticus in sleep (ESES)/continuous spikes and waves during slow sleep (CSWS) syndrome in children: An electroclinical evaluation according to the EEG patterns. Epilepsy Behav. 2016, 61, 107–111. [Google Scholar] [CrossRef]
- Shiraishi, H.; Haginoya, K.; Nakagawa, E.; Saitoh, S.; Kaneko, Y.; Nakasato, N.; Chan, D.; Otsubo, H. Magnetoencephalography localizing spike sources of atypical benign partial epilepsy. Brain Dev. 2014, 36, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nonclercq, A.; Foulon, M.; Verheulpen, D.; De Cock, C.; Buzatu, M.; Mathys, P.; Van Bogaert, P. Cluster-based spike detection algorithm adapts to interpatient and intrapatient variation in spike morphology. J. Neurosci. Methods 2012, 210, 259–265. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Y.; Li, Y.; Gao, Z.; Gai, Z.; Zhou, Y. SQNN: A spike-wave index quantification neural network with a pre-labeling algorithm for epileptiform activity identification and quantification in children. J. Neural Eng. 2022, 19, 016040. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Peters, J.M.; Hadjiloizou, S.; Prabhu, S.P.; Zarowski, M.; Stannard, K.M.; Takeoka, M.; Rotenberg, A.; Kothare, S.V.; Loddenkemper, T. Clinical staging and electroencephalographic evolution of continuous spikes and waves during sleep. Epilepsia 2012, 53, 1185–1195. [Google Scholar] [CrossRef]
- Cantalupo, G.; Pavlidis, E.; Beniczky, S.; Avanzini, P.; Gardella, E.; Larsson, P.G. Quantitative EEG analysis in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, S31–S40. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.E.; Peters, J.M.; Kothare, S.V.; Nordli, D.R., Jr.; Jensen, F.E.; Berg, A.T.; Loddenkemper, T. The tower of Babel: Survey on concepts and terminology in electrical status epilepticus in sleep and continuous spikes and waves during sleep in North America. Epilepsia 2013, 54, 741–750. [Google Scholar] [CrossRef]
- Carvalho, D.; Mendes, T.; Dias, A.I.; Leal, A. Interictal spike quantification in continuous spike-wave of sleep (CSWS): Clinical usefulness of a wearable EEG device. Epilepsy Behav. 2020, 104, 106902. [Google Scholar] [CrossRef]
- Poothrikovil, R.P.; Koul, R.L.; Mani, R.; Al Futaisi, A. Evolution of Ohtahara syndrome to continuous spikes and waves during slow sleep in an infant. Neurodiagn. J. 2012, 52, 261–274. [Google Scholar]
- Oguni, H.; Hirano, Y.; Nagata, S. Encephalopathy related to status epilepticus during slow sleep (ESES) as atypical evolution of Panayiotopoulos syndrome: An EEG and neuropsychological study. Epileptic Disord. 2020, 22, 67–72. [Google Scholar]
- Nava, E.; Mori, A.C.; Striano, P.; Ramantani, G. Atypical presentation of sunflower epilepsy featuring an EEG pattern of continuous spike waves during slow-wave sleep. Epileptic Disord. 2021, 23, 927–932. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.E.; Peters, J.M.; Harini, C.; Rotenberg, A.; Loddenkemper, T. Continuous spikes and waves during sleep: Electroclinical presentation and suggestions for management. Epilepsy Res. Treat. 2013, 2013, 583531. [Google Scholar] [CrossRef]
- Arzimanoglou, A.; Cross, H.J. Cognitive impairment and behavioral disorders in encephalopathy related to status epilepticus during slow sleep: Diagnostic assessment and outcome. Epileptic Disord. 2019, 21, S71–S75. [Google Scholar] [CrossRef]
- Margari, L.; Buttiglione, M.; Legrottaglie, A.R.; Presicci, A.; Craig, F.; Curatolo, P. Neuropsychiatric impairment in children with continuous spikes and waves during slow sleep: A long-term follow-up study. Epilepsy Behav. 2012, 25, 558–562. [Google Scholar] [CrossRef]
- Curnow, S.R.; Vogrin, S.J.; Barton, S.; Bailey, C.A.; Harvey, A.S. Focal cortical hypermetabolism in atypical benign rolandic epilepsy. Epilepsy Res. 2020, 161, 106288. [Google Scholar] [CrossRef]
- Bölsterli Heinzle, B.K.; Fattinger, S.; Kurth, S.; Lebourgeois, M.K.; Ringli, M.; Bast, T.; Critelli, H.; Schmitt, B.; Huber, R. Spike wave location and density disturb sleep slow waves in patients with CSWS (continuous spike waves during sleep). Epilepsia 2014, 55, 584–591. [Google Scholar] [CrossRef]
- Bölsterli, B.K.; Gardella, E.; Pavlidis, E.; Wehrle, F.M.; Tassinari, C.A.; Huber, R.; Rubboli, G. Remission of encephalopathy with status epilepticus (ESES) during sleep renormalizes regulation of slow wave sleep. Epilepsia 2017, 58, 1892–1901. [Google Scholar] [CrossRef]
- Oser, N.; Hubacher, M.; Nageleisen-Weiss, A.; van Mierlo, P.; Huber, R.; Weber, P.; Bölsterli, B.K.; Datta, A.N. 6-year course of sleep homeostasis in a case with epilepsy-aphasia spectrum disorder. Epilepsy Behav. Rep. 2021, 16, 100488. [Google Scholar] [CrossRef]
- Bölsterli Heinzle, B.K.; Bast, T.; Critelli, H.; Huber, R.; Schmitt, B. Age-dependency of location of epileptic foci in “continuous spike-and-waves during sleep”: A parallel to the posterior-anterior trajectory of slow wave activity. Neuropediatrics 2017, 48, 36–41. [Google Scholar]
- Filippini, M.; Arzimanoglou, A.; Gobbi, G. Neuropsychological approaches to epileptic encephalopathies. Epilepsia 2013, 54 (Suppl. 8), 38–44. [Google Scholar] [CrossRef]
- Halász, P.; Szűcs, A. Sleep and epilepsy link by plasticity. Front. Neurol. 2020, 11, 911. [Google Scholar] [CrossRef]
- Ng, R.; Hodges, E. Neurocognitive profiles of pediatric patients with ESES, generalized epilepsy, or focal epilepsy. Epilepsy Res. 2020, 167, 106351. [Google Scholar] [CrossRef]
- Panda, P.K.; Natarajan, V.; Sharawat, I.K. Neurocognitive profile of children with ESES, generalized, and focal epilepsy: Is there any difference? Epilepsy Res. 2021, 172, 106451. [Google Scholar] [CrossRef]
- Pesántez-Ríos, G.; Martínez-Bermejo, A.; Arcas, J.; Merino-Andreu, M.; Ugalde-Canitrot, A. Las evoluciones atipicas de la epilepsia rolandica son complicaciones predecibles [The atypical developments of rolandic epilepsy are predictable complications]. Rev. Neurol. 2015, 61, 106–113. [Google Scholar]
- Porat Rein, A.; Kramer, U.; Mitelpunkt, A. Development of ontology for self-limited epilepsy with centrotemporal spikes and application of data mining algorithms to identify new subtypes. Isr. Med. Assoc. J. 2019, 21, 503. [Google Scholar]
- Porat Rein, A.; Kramer, U.; Hausman Kedem, M.; Fattal-Valevski, A.; Mitelpunkt, A. Early prediction of encephalopathic transformation in children with benign epilepsy with centro-temporal spikes. Brain Dev. 2021, 43, 268–279. [Google Scholar] [CrossRef]
- Desprairies, C.; Dozières-Puyravel, B.; Ilea, A.; Bellavoine, V.; Nasser, H.; Delanöe, C.; Auvin, S. Early identification of epileptic encephalopathy with continuous spikes-and-waves during sleep: A case-control study. Eur. J. Paediatr. Neurol. 2018, 22, 837–844. [Google Scholar] [CrossRef]
- Caraballo, R.; Pavlidis, E.; Nikanorova, M.; Loddenkemper, T. Encephalopathy with continuous spike-waves during slow-wave sleep: Evolution and prognosis. Epileptic Disord. 2019, 21, S15–S21. [Google Scholar] [CrossRef]
- Aeby, A.; Santalucia, R.; Van Hecke, A.; Nebbioso, A.; Vermeiren, J.; Deconinck, N.; De Tiège, X.; Van Bogaert, P. A qualitative awake EEG score for the diagnosis of continuous spike and waves during sleep (CSWS) syndrome in self-limited focal epilepsy (SFE): A case-control study. Seizure 2021, 84, 34–39. [Google Scholar] [CrossRef]
- Peltola, M.E.; Palmu, K.; Liukkonen, E.; Gaily, E.; Vanhatalo, S. Semiautomatic quantification of spiking in patients with continuous spikes and waves in sleep: Sensitivity to settings and correspondence to visual assessment. Clin. Neurophysiol. 2012, 123, 1284–1290. [Google Scholar] [CrossRef]
- Peltola, M.E.; Sairanen, V.; Gaily, E.; Vanhatalo, S. Measuring spike strength in patients with continuous spikes and waves during sleep: Comparison of methods for prospective use as a clinical index. Clin. Neurophysiol. 2014, 125, 1639–1646. [Google Scholar] [CrossRef]
- Ouyang, G.; Wang, Y.; Yang, Z.; Li, X. Global synchronization of multichannel EEG in patients with electrical status epilepticus in sleep. Clin. EEG Neurosci. 2015, 46, 357–363. [Google Scholar] [CrossRef]
- Balaram, N.; Jose, J.; Gafoor, A.V.; Balachandran, S. Classification of electrical status epilepticus in sleep based on EEG patterns and spatiotemporal mapping of spikes. Epileptic Disord. 2022, 24, 1060–1072. [Google Scholar] [CrossRef]
- De Tiège, X.; Trotta, N.; Op de Beeck, M.; Bourguignon, M.; Marty, B.; Wens, V.; Nonclercq, A.; Goldman, S.; Van Bogaert, P. Neurophysiological activity underlying altered brain metabolism in epileptic encephalopathies with CSWS. Epilepsy Res. 2013, 105, 316–325. [Google Scholar] [CrossRef]
- Japaridze, N.; Muthuraman, M.; Dierck, C.; von Spiczak, S.; Boor, R.; Mideksa, K.G.; Anwar, R.A.; Deuschl, G.; Stephani, U.; Siniatchkin, M. Neuronal networks in epileptic encephalopathies with CSWS. Epilepsia 2016, 57, 1245–1255. [Google Scholar] [CrossRef]
- Magara, S.; Komatsubara, T.; Hojo, M.; Kobayashi, Y.; Yoshino, M.; Saitoh, A.; Tohyama, J. The association of epileptic focus estimated by magnetoencephalography with cognitive function in non-lesional epilepsy with continuous spikes and waves during slow wave sleep (ECSWS) children. Brain Dev. 2019, 41, 163–172. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Sun, J.; Niu, K.; Wang, P.; Xu, Y.; Wang, Y.; Chen, Q.; Zhang, K.; Wang, X. Relationship between brain activity, cognitive function, and sleep spiking activation in new-onset self-limited epilepsy with centrotemporal spikes. Front. Neurol. 2022, 13, 956838. [Google Scholar] [CrossRef]
- Moeller, F.; Moehring, J.; Ick, I.; Steinmann, E.; Wolff, S.; Jansen, O.; Boor, R.; Stephani, U.; Siniatchkin, M. EEG-fMRI in atypical benign partial epilepsy. Epilepsia 2013, 54, e103–e108. [Google Scholar] [CrossRef]
- Siniatchkin, M.; Van Bogaert, P. Pathophysiology of encephalopathy related to continuous spike and waves during sleep: The contribution of neuroimaging. Epileptic Disord. 2019, 21, S48–S53. [Google Scholar] [CrossRef]
- Toda, Y.; Kobayashi, K.; Hayashi, Y.; Inoue, T.; Oka, M.; Ohtsuka, Y. Effects of intravenous diazepam on high-frequency oscillations in EEGs with CSWS. Brain Dev. 2013, 35, 540–547. [Google Scholar] [CrossRef]
- Ohuchi, Y.; Akiyama, T.; Matsuhashi, M.; Kobayashi, K. High-frequency oscillations in a spectrum of pediatric epilepsies characterized by sleep-activated spikes in scalp EEG. Clin. Neurophysiol. 2019, 130, 1971–1980. [Google Scholar] [CrossRef]
- Cao, D.; Chen, Y.; Liao, J.; Nariai, H.; Li, L.; Zhu, Y.; Zhao, X.; Hu, Y.; Wen, F.; Zhai, Q. Scalp EEG high frequency oscillations as a biomarker of treatment response in epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). Seizure 2019, 71, 151–157. [Google Scholar] [CrossRef]
- Gong, P.; Yang, Z.X.; Xue, J.; Qian, P.; Yang, H.P.; Liu, X.Y.; Bian, K.G. Application of scalp-recorded high-frequency oscillations in epileptic encephalopathy with continuous spike-and-wave during sleep. J. Peking Univ. Health Sci. 2018, 50, 213–220. [Google Scholar]
- Gong, P.; Xue, J.; Qian, P.; Yang, H.; Liu, X.; Cai, L.; Bian, K.; Yang, Z. Scalp-recorded high-frequency oscillations in childhood epileptic encephalopathy with continuous spike-and-wave during sleep with different etiologies. Brain Dev. 2018, 40, 299–310. [Google Scholar] [CrossRef]
- Giacomini, T.; Luria, G.; D’Amario, V.; Croci, C.; Cataldi, M.; Piai, M.; Nobile, G.; Bruni, O.; Consales, A.; Mancardi, M.M.; et al. On the role of REM sleep microstructure in suppressing interictal spikes in electrical status epilepticus during sleep. Clin. Neurophysiol. 2022, 136, 62–68. [Google Scholar] [CrossRef]
- Filippini, M.; Boni, A.; Giannotta, M.; Pini, A.; Russo, A.; Musti, M.A.; Guerra, A.; Lassonde, M.; Gobbi, G. Comparing cortical auditory processing in children with typical and atypical benign epilepsy with centrotemporal spikes: Electrophysiologic evidence of the role of non-rapid eye movement sleep abnormalities. Epilepsia 2015, 56, 726–734. [Google Scholar] [CrossRef]
- Giovanardi Rossi, P.; Parmeggiani, A.; Posar, A.; Scaduto, M.C.; Chiodo, S.; Vatti, G. Landau-Kleffner syndrome (LKS): Long-term follow-up and links with electrical status epilepticus during sleep (ESES). Brain Dev. 1999, 21, 90–98. [Google Scholar] [CrossRef]
- Arican, P.; Gencpinar, P.; Olgac Dundar, N.; Tekgul, H. Electrical status epilepticus during slow-wave sleep (ESES): Current perspectives. J. Pediatr. Neurosci. 2021, 16, 91–96. [Google Scholar]
- Zhang, K.; Yan, Y.; Su, T. Treatment strategies for encephalopathy related to status epilepticus during slow sleep, a narrative review of the literature. Rev. Neurosci. 2020, 31, 793–802. [Google Scholar] [CrossRef]
- Carreño, M.; Fernández, S. Sleep-related epilepsy. Curr. Treat. Options Neurol. 2016, 18, 23. [Google Scholar] [CrossRef]
- Raha, S.; Shah, U.; Udani, V. Neurocognitive and neurobehavioral disabilities in epilepsy with electrical status epilepticus in slow sleep (ESES) and related syndromes. Epilepsy Behav. 2012, 25, 381–385. [Google Scholar] [CrossRef]
- McTague, A.; Cross, J.H. Treatment of epileptic encephalopathies. CNS Drugs 2013, 27, 175–184. [Google Scholar] [CrossRef]
- Hani, A.J.; Mikati, M.A. Current and emerging therapies of severe epileptic encephalopathies. Semin. Pediatr. Neurol. 2016, 23, 180–186. [Google Scholar] [CrossRef]
- van den Munckhof, B.; Alderweireld, C.; Davelaar, S.; van Teeseling, H.C.; Nikolakopoulos, S.; Braun, K.P.J.; Jansen, F.E. Treatment of electrical status epilepticus in sleep: Clinical and EEG characteristics and response to 147 treatments in 47 patients. Eur. J. Paediatr. Neurol. 2018, 22, 64–71. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Chapman, K.; Peters, J.M.; Klehm, J.; Jackson, M.C.; Berg, A.T.; Loddenkemper, T. Treatment for continuous spikes and waves during sleep (CSWS): Survey on treatment choices in North America. Epilepsia 2014, 55, 1099–1108. [Google Scholar] [CrossRef]
- Kessi, M.; Yan, F.; Pan, L.; Chen, B.; Olatoutou, E.; Li, D.; He, F.; Rugambwa, T.; Yang, L.; Peng, J.; et al. Treatment for the benign childhood epilepsy with centrotemporal spikes: A monocentric study. Front. Neurol. 2021, 12, 670958. [Google Scholar] [CrossRef]
- van den Munckhof, B.; van Dee, V.; Sagi, L.; Caraballo, R.H.; Veggiotti, P.; Liukkonen, E.; Loddenkemper, T.; Sánchez Fernández, I.; Buzatu, M.; Bulteau, C.; et al. Treatment of electrical status epilepticus in sleep: A pooled analysis of 575 cases. Epilepsia 2015, 56, 1738–1746. [Google Scholar] [CrossRef]
- Zhang, J. The effectiveness and safety of hormonal combinations of antiepileptic drugs in the treatment of epileptic electrical continuity in children during sleep: A meta-analysis. Comput. Intell. Neurosci. 2022, 2022, 5395383. [Google Scholar] [CrossRef] [PubMed]
- Moresco, L.; Bruschettini, M.; Calevo, M.G.; Siri, L. Pharmacological treatment for continuous spike-wave during slow wave sleep syndrome and Landau-Kleffner syndrome. Cochrane Database Syst. Rev. 2020, 11, CD013132. [Google Scholar] [CrossRef]
- Kanemura, H.; Sano, F.; Sugita, K.; Aihara, M. Effects of levetiracetam on seizure frequency and neuropsychological impairments in children with refractory epilepsy with secondary bilateral synchrony. Seizure 2013, 22, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, F.; Jiang, L.; Hu, Y.; Feng, C. Levetiracetam efficacy in children with epilepsy with electrical status epilepticus in sleep. Epilepsy Behav. 2015, 44, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Su, T.F.; Xu, S.Q.; Chen, L. Efficacy of levetiracetam combined with short-term clonazepam in treatment of electrical status epilepticus during sleep in children with benign childhood epilepsy with centrotemporal spikes. Chin. J. Contemp. Pediatr. 2014, 16, 829–833. [Google Scholar]
- Wiwattanadittakul, N.; Depositario-Cabacar, D.; Zelleke, T.G. Electrical status epilepticus in sleep (ESES)—Treatment pattern and EEG outcome in children with very high spike-wave index. Epilepsy Behav. 2020, 105, 106965. [Google Scholar] [CrossRef]
- Yuan, Q.; Li, F.; Zhong, H. Early diagnosis, treatment and prognosis of epilepsy with continuous spikes and waves during slow sleep. Int. J. Clin. Exp. Med. 2015, 8, 4052–4058. [Google Scholar]
- Vrielynck, P.; Marique, P.; Ghariani, S.; Lienard, F.; de Borchgrave, V.; van Rijckevorsel, K.; Bonnier, C. Topiramate in childhood epileptic encephalopathy with continuous spike-waves during sleep: A retrospective study of 21 cases. Eur. J. Paediatr. Neurol. 2017, 21, 305–311. [Google Scholar] [CrossRef]
- Kanemura, H.; Sano, F.; Hoshino, H.; Takayama, K.; Aihara, M. Effects of perampanel on secondary bilateral synchrony and behavioral problems in adolescents with epilepsy showing insufficient response with levetiracetam. Seizure 2020, 80, 131–137. [Google Scholar] [CrossRef]
- Yu, T.; Teng, Z.-T.; Liu, X.-Y.; Wang, H. Effectiveness of perampanel in the treatment of pediatric patients with focal epilepsy and ESES: A single-center retrospective study. Front. Pharmacol. 2022, 13, 1026836. [Google Scholar] [CrossRef]
- Li, D.; Huang, S.; Wang, X.; Yang, L.; Song, T. Efficacy and adverse reactions of perampanel in the treatment of epilepsy in children. Front. Neurol. 2022, 13, 924057. [Google Scholar] [CrossRef]
- Grosso, S.; Parisi, P.; Giordano, L.; di Bartolo, R.; Balestri, P. Lacosamide efficacy in epileptic syndromes with continuous spike and waves during slow sleep (CSWS). Epilepsy Res. 2014, 108, 1604–1608. [Google Scholar] [CrossRef] [PubMed]
- Fatema, K.; Rahman, M.M.; Begum, S. Characteristics and management of children with continuous spikes and waves during slow sleep. Mymensingh Med. J. 2015, 24, 806–812. [Google Scholar] [PubMed]
- Fejerman, N.; Caraballo, R.; Cersósimo, R.; Ferraro, S.M.; Galicchio, S.; Amartino, H. Sulthiame add-on therapy in children with focal epilepsies associated with encephalopathy related to electrical status epilepticus during slow sleep (ESES). Epilepsia 2012, 53, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Kanmaz, S.; Simsek, E.; Serin, H.M.; Yilmaz, S.; Aktan, G.; Tekgul, H.; Gokben, S. Sulthiame add-on treatment in children with epileptic encephalopathy with status epilepticus: An efficacy analysis in etiologic subgroups. Neurol. Sci. 2021, 42, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Topçu, Y.; Kılıç, B.; Tekin, H.G.; Aydın, K.; Turanlı, G. Effects of sulthiame on seizure frequency and EEG in children with electrical status epilepticus during slow sleep. Epilepsy Behav. 2021, 116, 107793. [Google Scholar] [CrossRef] [PubMed]
- Fine, A.L.; Wirrell, E.C.; Wong-Kisiel, L.C.; Nickels, K.C. Acetazolamide for electrical status epilepticus in slow-wave sleep. Epilepsia 2015, 56, e134–e138. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, F.; Jiang, L.; Hu, Y.; Feng, C. A prospective study of dexamethasone therapy in refractory epileptic encephalopathy with continuous spike-and-wave during sleep. Epilepsy Behav. 2016, 55, 1–5. [Google Scholar] [CrossRef]
- Bast, T.; Richter, S.; Ebinger, F.; Rating, D.; Wiemer-Kruel, A.; Schubert-Bast, S. Efficacy and tolerability of methylprednisolone pulse therapy in childhood epilepsies other than infantile spasms. Neuropediatrics 2014, 45, 378–385. [Google Scholar]
- Hempel, A.; Frost, M.; Agarwal, N. Language and behavioral outcomes of treatment with pulse-dose prednisone for electrical status epilepticus in sleep (ESES). Epilepsy Behav. 2019, 94, 93–99. [Google Scholar] [CrossRef]
- Jauhari, P.; Madaan, P.; Sirolia, V.; Sharma, S.; Chakrabarty, B.; Gulati, S. Neurobehavioral deterioration associated with sleep-augmented epileptiform abnormalities: A steroid responsive state in children. Epilepsy Behav. 2022, 129, 108505. [Google Scholar] [CrossRef]
- Meng, L.P.; Dai, Y.Y. A clinical analysis of electrical status epilepticus during sleep in children and a follow-up study of methylprednisolone pulse therapy. Chin. J. Contemp. Pediatr. 2019, 21, 348–353. [Google Scholar]
- Chen, J.; Yang, Z.; Liu, X.; Ji, T.; Fu, N.; Wu, Y.; Xiong, H.; Wang, S.; Chang, X.; Zhang, Y.; et al. Efficacy of methylprednisolone therapy for electrical status epilepticus during sleep in children. Chin. J. Pediatr. 2014, 52, 678–682. [Google Scholar]
- Baumer, F.M.; McNamara, N.A.; Fine, A.L.; Pestana-Knight, E.; Shellhaas, R.A.; He, Z.; Arndt, D.H.; Gaillard, W.D.; Kelley, S.A.; Nagan, M.; et al. Treatment practices and outcomes in continuous spike and wave during slow wave sleep: A multicenter collaboration. J. Pediatr. 2021, 232, 220–228.e3. [Google Scholar] [CrossRef]
- Kılıç, B.; Acar, M.; Topçu, Y.; Turanlı, G. Epileptic encephalopathy with electrical status epilepticus during slow sleep: Evaluation of treatment response from a tertiary center. Turk. J. Pediatr. 2022, 64, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Uliel-Sibony, S.; Kramer, U. Benign childhood epilepsy with centro-temporal spikes (BCECTSs), electrical status epilepticus in sleep (ESES), and academic decline—How aggressive should we be? Epilepsy Behav. 2015, 44, 117–120. [Google Scholar] [CrossRef]
- Jyonouchi, H.; Geng, L. Resolution of EEG findings and clinical improvement in a patient with encephalopathy and ESES with a combination of immunomodulating agents other than corticosteroids: A case report. Epilepsy Behav. Rep. 2020, 14, 100379. [Google Scholar] [CrossRef] [PubMed]
- Reyes, G.; Flesler, S.; Armeno, M.; Fortini, S.; Ariela, A.; Cresta, A.; Mestre, G.; Caraballo, R.H. Ketogenic diet in patients with epileptic encephalopathy with electrical status epilepticus during slow sleep. Epilepsy Res. 2015, 113, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.A.; Kossoff, E.H. How effective is the ketogenic diet for electrical status epilepticus of sleep? Epilepsy Res. 2016, 127, 339–343. [Google Scholar] [CrossRef]
- Ville, D.; Chiron, C.; Laschet, J.; Dulac, O. The ketogenic diet can be used successfully in combination with corticosteroids for epileptic encephalopathies. Epilepsy Behav. 2015, 48, 61–65. [Google Scholar] [CrossRef]
- Pasca, L.; Caraballo, R.H.; De Giorgis, V.; Reyes, J.G.; Macasaet, J.A.; Masnada, S.; Armeno, M.; Musicco, M.; Tagliabue, A.; Veggiotti, P. Ketogenic diet use in children with intractable epilepsy secondary to malformations of cortical development: A two-centre experience. Seizure 2018, 57, 34–37. [Google Scholar] [CrossRef]
- Wilson, R.B.; Eliyan, Y.; Sankar, R.; Hussain, S.A. Amantadine: A new treatment for refractory electrical status epilepticus in sleep. Epilepsy Behav. 2018, 84, 74–78. [Google Scholar] [CrossRef]
- Carosella, C.M.; Greiner, H.M.; Byars, A.W.; Arthur, T.M.; Leach, J.L.; Turner, M.; Holland, K.D.; Mangano, F.T.; Arya, R. Vagus nerve stimulation for electrographic status epilepticus in slow-wave sleep. Pediatr. Neurol. 2016, 60, 66–70. [Google Scholar] [CrossRef]
- Faria, P.; Fregni, F.; Sebastião, F.; Dias, A.I.; Leal, A. Feasibility of focal transcranial DC polarization with simultaneous EEG recording: Preliminary assessment in healthy subjects and human epilepsy. Epilepsy Behav. 2012, 25, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, P. Current status of treatments for children with electrical status in slow-wave sleep (ESES/CSWS). Epilepsy Curr. 2017, 17, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Willis, E.; Sharp, G.B. Absence status after starting clobazam in a patient with syndrome of continuous spike and wave during slow sleep (CSWS). Neurol. India 2014, 62, 685–687. [Google Scholar] [CrossRef]
- Liu, M.; Ji, T.; Liu, Q.; Wang, S.; Wu, Y.; Wang, W.; Cheng, W.; Wang, R.; Yu, G.; Liu, X.; et al. Generalized seizures presurgically in a cohort of children with hemispherectomy: Predictors and a potential link to surgical outcome? Seizure 2018, 58, 101–109. [Google Scholar] [CrossRef]
- Ng, Y.-T. Increasing blurriness of the borders between focal and generalized as well as cryptogenic and idiopathic epilepsies in defining the role for focal epilepsy surgery. Semin. Pediatr. Neurol. 2014, 21, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Gröppel, G.; Dorfer, C.; Dressler, A.; Mühlebner, A.; Porsche, B.; Hainfellner, J.A.; Czech, T.; Feucht, M. Immediate termination of electrical status epilepticus in sleep after hemispherotomy is associated with significant progress in language development. Dev. Med. Child Neurol. 2017, 59, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Weil, A.G.; Ibrahim, G.M.; Fallah, A.; Korman, B.; Ragheb, J.; Bhatia, S.; Duchowny, M. Surgical management of pediatric patients with encephalopathy due to electrical status epilepticus during sleep (ESES). Epileptic Disord. 2020, 22, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Marashly, A.; Koop, J.; Loman, M.; Lee, Y.-W.; Lew, S.M. Examining the utility of resective epilepsy surgery in children with electrical status epilepticus in sleep: Long term clinical and electrophysiological outcomes. Front. Neurol. 2020, 10, 1397. [Google Scholar] [CrossRef] [PubMed]
- Alawadhi, A.; Appendino, J.P.; Hader, W.; Rosenblatt, B.; Moreau, J.T.; Dubeau, F.; Dudley, R.W.R.; Myers, K.A. Surgically remediable secondary network epileptic encephalopathies with continuous spike wave in sleep: Lesions may not be visible on brain magnetic resonance imaging (MRI). J. Child Neurol. 2022, 37, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Jeong, A.; Strahle, J.; Vellimana, A.K.; Limbrick, D.D., Jr.; Smyth, M.D.; Bertrand, M. Hemispherotomy in children with electrical status epilepticus of sleep. J. Neurosurg. Pediatr. 2017, 19, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Yokosako, S.; Muraoka, N.; Watanabe, S.; Kosugi, K.; Takayama, Y.; Iijima, K.; Kimura, Y.; Kaneko, Y.; Sumitomo, N.; Saito, T.; et al. Corpus callosotomy in pediatric patients with non-lesional epileptic encephalopathy with electrical status epilepticus during sleep. Epilepsy Behav. Rep. 2021, 16, 100463. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Serdaroglu, G.; Akcay, A.; Gokben, S. Clinical characteristics and outcome of children with electrical status epilepticus during slow wave sleep. J. Pediatr. Neurosci. 2014, 9, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.C.; Brazzo, D.; Altieri, N.; Balottin, U.; Veggiotti, P. Long-term evolution of neuropsychological competences in encephalopathy with status epilepticus during sleep: A variable prognosis. Epilepsia 2013, 54 (Suppl. 7), 77–85. [Google Scholar] [CrossRef]
- Hegyi, M.; Siegler, Z.; Fogarasi, A.; Barsi, P.; Halász, P. Long term follow-up of lesional and non-lesional patients with electrical status epilepticus in slow wave sleep. Ideggyogy Sz. 2016, 69, 21–28. [Google Scholar] [CrossRef]
- De Giorgis, V.; Filippini, M.; Macasaet, J.A.; Masnada, S.; Veggiotti, P. Neurobehavioral consequences of continuous spike and waves during slow sleep (CSWS) in a pediatric population: A pattern of developmental hindrance. Epilepsy Behav. 2017, 74, 1–9. [Google Scholar] [CrossRef]
- Escobar Fernández, L.; Coccolo Góngora, A.; Vázquez López, M.; Polo Arrondo, A.P.; Miranda Herrero, M.C.; Barredo Valderrama, E.; Castro de Castro, P. Patrón punta-onda continua en el sueño lento: Nuestra experiencia durante 20 años [Continuous spike-waves during slow-wave sleep: Experience during 20 years]. An. Pediatr. (Engl. Ed.) 2019, 91, 180–188. [Google Scholar] [CrossRef]
- Saraf, U.U.; Asranna, A.; Menon, R.N.; Mohan, P.M.; Vipina, V.P.; Radhakrishnan, A.; Cherian, A.; Thomas, S.V. Predictors of one-year language and seizure outcomes in children with epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). Seizure 2020, 81, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Öztoprak, Ü.; Yayici Köken, Ö.; Aksoy, E.; Yüksel, D. Spike-wave index assessment and electro-clinical correlation in patients with encephalopathy associated with epileptic state during slow sleep (ESES/CSWS); Single-center experience. Epilepsy Res. 2021, 170, 106549. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Cantalupo, G.; Larsson, P.G.; Fontana, E.; Bernardina, B.D.; Rubboli, G.; Darra, F. EEG features in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, D.T.; Stanford, L.D. Serial neuropsychological testing before and after hemispherectomy in a child with electrical status epilepticus in slow wave sleep. Epilepsy Behav. Rep. 2022, 18, 100539. [Google Scholar] [CrossRef] [PubMed]
- Ucar, H.K.; Arhan, E.; Aydin, K.; Hirfanoglu, T.; Serdaroglu, A. Electrical status epilepticus during sleep (ESES) in benign childhood epilepsy with centrotemporal spikes (BCECTS): Insights into predictive factors, and clinical and EEG outcomes. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1885–1896. [Google Scholar] [PubMed]
- Tuft, M.; Årva, M.; Bjørnvold, M.; Wilson, J.A.; Nakken, K.O. Landau-Kleffner syndrome. Tidsskr. Nor. Laegeforen. 2015, 135, 2061–2064. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.Y.; Ke, D.S.; Chaou, W.T. Landau-Kleffner syndrome: An acquired epileptic aphasia. Acta Neurol. 2015, 24, 34–35. [Google Scholar] [PubMed]
- Muzio, M.R.; Cascella, M.; Al Khalili, Y. Landau-Kleffner Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chowdhury, N.; Bansal, A.R.; Goyal, R.; Nikhila, G. Cerebral dominance in an unusual case of Landau-Kleffner syndrome. BMJ Case Rep. 2021, 14, e246696. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Cejas, N.; Chamorro, N.; Kaltenmeier, M.C.; Fortini, S.; Soprano, A.M. Landau-Kleffner syndrome: A study of 29 patients. Seizure 2014, 23, 98–104. [Google Scholar] [CrossRef]
- Uğur, C.; Saday Duman, N.; Bektaş, O.; Kağan Gürkan, C. Antiepileptic treatment in a child with Landau Kleffner syndrome: A case report. Turk Psikiyatr. Derg. 2014, 25, 282–286. [Google Scholar]
- Riccio, C.A.; Vidrine, S.M.; Cohen, M.J.; Acosta-Cotte, D.; Park, Y. Neurocognitive and behavioral profiles of children with Landau-Kleffner syndrome. Appl. Neuropsychol. Child 2017, 6, 345–354. [Google Scholar] [CrossRef]
- Besag, F.M.C.; Vasey, M.J. Social cognition and psychopathology in childhood and adolescence. Epilepsy Behav. 2019, 100, 106210. [Google Scholar] [CrossRef]
- Ewen, J.B.; Marvin, A.R.; Law, K.; Lipkin, P.H. Epilepsy and autism severity: A study of 6975 children. Autism Res. 2019, 12, 1251–1259. [Google Scholar] [CrossRef]
- Sigafoos, J.; O’Reilly, M.F.; Ledbetter-Cho, K.; Lim, N.; Lancioni, G.E.; Marschik, P.B. Addressing sequelae of developmental regression associated with developmental disabilities: A systematic review of behavioral and educational intervention studies. Neurosci. Biobehav. Rev. 2019, 96, 56–71. [Google Scholar] [CrossRef]
- Peng, B.-W.; Zhu, H.-X.; Wang, X.-Y.; Li, X.-J.; Liang, H.-C.; Li, J.-L.; Zhang, F.-Q.; Ning, S.-Y.; Zhong, Y.-Y.; Chen, W.-X. A follow-up study in children with status epilepticus during sleep: From clinical spectrum to outcome. Epilepsy Behav. 2021, 117, 107843. [Google Scholar] [CrossRef]
- Ahmed, M.; Saleem, A.; Nasir, S.; Ariff, M.; Iftikhar, P. Landau-Kleffner syndrome: A diagnostic challenge. Cureus 2020, 12, e7182. [Google Scholar] [CrossRef]
- Lévêque, Y.; Roulet-Perez, E.; Deonna, T.; Moulin, A.; Fornoni, L.; Mayor-Dubois, C.; Caclin, A.; Tillmann, B. Music processing deficits in Landau-Kleffner syndrome: Four case studies in adulthood. Cortex 2020, 129, 99–111. [Google Scholar] [CrossRef]
- Kaga, M.; Inagaki, M.; Ohta, R. Epidemiological study of Landau-Kleffner syndrome (LKS) in Japan. Brain Dev. 2014, 36, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Plyler, E.; Harkrider, A.W. Serial auditory-evoked potentials in the diagnosis and monitoring of a child with Landau-Kleffner syndrome. J. Am. Acad. Audiol. 2013, 24, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Motwani, N.; Afsar, S.; Dixit, N.S.; Sharma, N. Landau-Kleffner syndrome: An uncommon dealt with case in Southeast Asia. BMJ Case Rep. 2015, 2015, bcr2015212333. [Google Scholar] [CrossRef]
- Klimova, B.; Valis, M.; Hort, J.; Kuca, K. Selected rare paediatric communication neurological disorders. J. Appl. Biomed. 2019, 17, 33. [Google Scholar] [CrossRef]
- De Lima, T.A.; Zuanetti, P.A.; Nunes, M.E.N.; Hamad, A.P.A. Differential diagnosis between autism spectrum disorder and other developmental disorders with emphasis on the preschool period. World J. Pediatr. 2023, 19, 715–726. [Google Scholar] [CrossRef]
- Murugesan, B.G.; Jafroodifar, A.; Anilkumar, A.C.; Leontieva, L. Differential diagnosis of Landau-Kleffner syndrome versus post encephalitis syndrome in a 13-year-old boy with autism spectrum disorder. Cureus 2020, 12, e9385. [Google Scholar] [CrossRef] [PubMed]
- Rezayi, A.; Feshangchi-Bonab, M.; Taherian, R. An uncommon presentation of mucopolysaccharidosis type IIIb. Iran. J. Child Neurol. 2019, 13, 105–111. [Google Scholar] [PubMed]
- Van Bogaert, P.; King, M.D.; Paquier, P.; Wetzburger, C.; Labasse, C.; Dubru, J.M.; Deonna, T. Acquired auditory agnosia in childhood and normal sleep electroencephalography subsequently diagnosed as Landau-Kleffner syndrome: A report of three cases. Dev. Med. Child Neurol. 2013, 55, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Duffy, F.H.; Eksioglu, Y.Z.; Rotenberg, A.; Madsen, J.R.; Shankardass, A.; Als, H. The frequency modulated auditory evoked response (FMAER), a technical advance for study of childhood language disorders: Cortical source localization and selected case studies. BMC Neurol. 2013, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Nieh, S.E.; Sherr, E.H. Epileptic encephalopathies: New genes and new pathways. Neurotherapeutics 2014, 11, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Vears, D.F.; Turner, S.J.; Smith, R.L.; Berkovic, S.F.; Sadleir, L.G.; Scheffer, I.E. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia 2013, 54, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Zavadenko, N.N.; Kholin, A.A.; Zavadenko, A.N.; Michurina, E.S. Narusheniia razvitiiarechi pri épilepsii: Patofiziologicheskie mekhanizmy i terapevticheskie podkhody [Speech and language neurodevelopmental disorders in epilepsy: Pathophysiologic mechanisms and therapeutic approaches]. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova 2018, 118, 118–125. [Google Scholar] [CrossRef]
- Van Bogaert, P. Epileptic encephalopathy with continuous spike-waves during slow-wave sleep including Landau-Kleffner syndrome. Handb. Clin. Neurol. 2013, 111, 635–640. [Google Scholar]
- Nemati, R.; Nabipour, I.; Javadi, H.; Chabi, N.; Assadi, M. Regional cerebral blood-flow with 99mTc-ECD brain perfusion SPECT in Landau-Kleffner syndrome: Report of two cases. Case Rep. Radiol. 2014, 2014, 617343. [Google Scholar] [CrossRef] [PubMed]
- Pullens, P.; Pullens, W.; Blau, V.; Sorger, B.; Jansma, B.M.; Goebel, R. Evidence for normal letter-sound integration, but altered language pathways in a case of recovered Landau-Kleffner syndrome. Brain Cogn. 2015, 99, 32–45. [Google Scholar] [CrossRef]
- Kawai, M.; Abe, Y.; Yumoto, M.; Kubota, M. Aphasia and a dual-stream language model in a 4-year-old female with Landau-Kleffner syndrome. Neuropediatrics 2022, 53, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.N.; Oser, N.; Ramelli, G.P.; Gobbin, N.Z.; Lantz, G.; Penner, I.K.; Weber, P. BECTS evolving to Landau-Kleffner syndrome and back by subsequent recovery: A longitudinal language reorganization case study using fMRI, source EEG, and neuropsychological testing. Epilepsy Behav. 2013, 27, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Jokel, R.; Meloff, K. Acquired epileptiform aphasia: 44 years after diagnosis. Epilepsy Behav. Rep. 2020, 14, 100388. [Google Scholar] [CrossRef] [PubMed]
- Cockerell, I.; Bølling, G.; Nakken, K.O. Landau-Kleffner syndrome in Norway: Long-term prognosis and experiences with the health services and educational systems. Epilepsy Behav. 2011, 21, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fainberg, N.; Harper, A.; Tchapyjnikov, D.; Mikati, M.A. Response to immunotherapy in a patient with Landau-Kleffner syndrome and GRIN2A mutation. Epileptic Disord. 2016, 18, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Goldberg, R.; Miles, D.; Bojko, A.; Riviello, J., Jr. Episodic epileptic verbal auditory agnosia in Landau Kleffner syndrome treated with combination diazepam and corticosteroids. J. Child Neurol. 2014, 29, 1291–1298. [Google Scholar] [CrossRef]
- Grote, C.L.; Van Slyke, P.; Hoeppner, J.-A.B. Language outcome following multiple subpial transection for Landau-Kleffner syndrome. Brain 1999, 122, 561–566. [Google Scholar] [CrossRef]
- Besag, F.; Caplan, R.; Aldenkamp, A.; Dunn, D.W.; Gobbi, G.; Sillanpää, M. Psychiatric and behavioural disorders in children with epilepsy (ILAE Task Force Report): Behavioural effects of epilepsy surgery. Epileptic Disord. 2016, 18 (Suppl. S1), S68–S76. [Google Scholar] [CrossRef]
- Downes, M.; Greenaway, R.; Clark, M.; Helen Cross, J.; Jolleff, N.; Harkness, W.; Kaliakatsos, M.; Boyd, S.; White, S.; Neville, B.G.R. Outcome following multiple subpial transection in Landau-Kleffner syndrome and related regression. Epilepsia 2015, 56, 1760–1766. [Google Scholar] [CrossRef]
- Fine, A.; Nickels, K. Temporoparietal resection in a patient with Landau-Kleffner syndrome. Semin. Pediatr. Neurol. 2014, 21, 96–100. [Google Scholar] [CrossRef]
- Hajtovic, S.; LoPresti, M.A.; Zhang, L.; Katlowitz, K.A.; Kizek, D.J.; Lam, S. The role of vagus nerve stimulation in genetic etiologies of drug-resistant epilepsy: A meta-analysis. J. Neurosurg. Pediatr. 2022, 29, 667–680. [Google Scholar] [CrossRef]
- van der Meulen, I.; Pangalila, R.F.; van de Sandt-Koenderman, W.M.E. Cognitive linguistic treatment in Landau Kleffner syndrome: Improvement in daily life communication. Child Neurol. Open 2021, 8, 2329048X211022196. [Google Scholar] [PubMed]
- Pangalila, R.; van der Meulen, I. What is the effect of pharmacological treatment for continuous spike-wave during slow wave sleep syndrome and Landau-Kleffner syndrome? A Cochrane review summary with commentary. Dev. Med. Child Neurol. 2022, 64, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Cersósimo, R.O.; Fortini, P.S.; Ornella, L.; Buompadre, M.C.; Vilte, C.; Princich, J.P.; Fejerman, N. Congenital hemiparesis, unilateral polymicrogyria and epilepsy with or without status epilepticus during sleep: A study of 66 patients with long-term follow-up. Epileptic Disord. 2013, 15, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Q.; Zhang, W.-N.; Hu, L.-Y.; Liu, M.-J.; Zou, L.-P. Syndrome of electrical status epilepticus during sleep: Epileptic encephalopathy related to brain development. Pediatr. Neurol. 2016, 56, 35–41. [Google Scholar] [CrossRef]
- Sun, Q.-Q.; Zhou, C.; Yang, W.; Petrus, D. Continuous spike-waves during slow-wave sleep in a mouse model of focal cortical dysplasia. Epilepsia 2016, 57, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.B.; Magalhães, S.C.; Pessoa, A.; Bueno, C.; Masruha, M.R.; Sobreira-Neto, M.A. Electrical status epilepticus during sleep in patients with congenital Zikavirus syndrome: An unprecedented clinical finding. Seizure 2020, 81, 250–253. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, H.; Liu, Z.; Wu, Q. Electrical status epilepticus in sleep affects intrinsically connected networks in patients with benign childhood epilepsy with centrotemporal spikes. Epilepsy Behav. 2020, 106, 107032. [Google Scholar] [CrossRef]
- Issa, N.P. Neurobiology of continuous spike-wave in slow-wave sleep and Landau-Kleffner syndromes. Pediatr. Neurol. 2014, 51, 287–296. [Google Scholar] [CrossRef]
- Gibbs, S.A.; Nobili, L.; Halász, P. Interictal epileptiform discharges in sleep and the role of the thalamus in encephalopathy related to status epilepticus during slow sleep. Epileptic Disord. 2019, 21, S54–S61. [Google Scholar] [CrossRef]
- Losito, E.; Battaglia, D.; Chieffo, D.; Raponi, M.; Ranalli, D.; Contaldo, I.; Giansanti, C.; De Clemente, V.; Quintiliani, M.; Antichi, E.; et al. Sleep-potentiated epileptiform activity in early thalamic injuries: Study in a large series (60 cases). Epilepsy Res. 2015, 109, 90–99. [Google Scholar] [CrossRef]
- Leal, A.; Calado, E.; Vieira, J.P.; Mendonça, C.; Ferreira, J.C.; Ferreira, H.; Carvalho, D.; Furtado, F.; Gomes, R.; Monteiro, J.P. Anatomical and physiological basis of continuous spike-wave of sleep syndrome after early thalamic lesions. Epilepsy Behav. 2018, 78, 243–255. [Google Scholar] [CrossRef]
- Kersbergen, K.J.; de Vries, L.S.; Leijten, F.S.S.; Braun, K.P.J.; Nievelstein, R.A.J.; Groenendaal, F.; Benders, M.J.N.L.; Jansen, F.E. Neonatal thalamic hemorrhage is strongly associated with electrical status epilepticus in slow wave sleep. Epilepsia 2013, 54, 733–740. [Google Scholar] [CrossRef]
- Öztürk, Z.; Karalok, Z.S.; Güneş, A. Reduced thalamic volume is strongly associated with electrical status epilepticus in sleep. Acta Neurol. Belg. 2021, 121, 211–217. [Google Scholar] [CrossRef]
- Bartolini, E.; Falchi, M.; Zellini, F.; Parrini, E.; Grisotto, L.; Cosottini, M.; Posar, A.; Parmeggiani, A.; Ambrosetto, G.; Ferrari, A.R.; et al. The syndrome of polymicrogyria, thalamic hypoplasia, and epilepsy with CSWS. Neurology 2016, 86, 1250–1259. [Google Scholar] [CrossRef]
- van den Munckhof, B.; Zwart, A.F.; Weeke, L.C.; Claessens, N.H.P.; Plate, J.D.J.; Leemans, A.; Kuijf, H.J.; van Teeseling, H.C.; Leijten, F.S.S.; Benders, M.J.N.; et al. Perinatal thalamic injury: MRI predictors of electrical status epilepticus in sleep and long-term neurodevelopment. Neuroimage Clin. 2020, 26, 102227. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Peters, J.M.; Akhondi-Asl, A.; Klehm, J.; Warfield, S.K.; Loddenkemper, T. Reduced thalamic volume in patients with electrical status epilepticus in sleep. Epilepsy Res. 2017, 130, 74–80. [Google Scholar] [CrossRef]
- Agarwal, R.; Kumar, A.; Tiwari, V.N.; Chugani, H. Thalamic abnormalities in children with continuous spike-wave during slow-wave sleep: An F-18-fluorodeoxyglucose positron emission tomography perspective. Epilepsia 2016, 57, 263–271. [Google Scholar] [CrossRef]
- Kilic, H.; Yilmaz, K.; Asgarova, P.; Kizilkilic, O.; Hatay, G.H.; Ozturk-Isik, E.; Yalcinkaya, C.; Saltik, S. Electrical status epilepticus in sleep: The role of thalamus in etiopathogenesis. Seizure 2021, 93, 44–50. [Google Scholar] [CrossRef]
- Leal, A. Spatial and temporal dynamics of epileptic activity at sleep onset in the encephalopathy with status epilepticus during slow sleep (ESES) after unilateral thalamic lesions. Clin. Neurophysiol. 2021, 132, 114–125. [Google Scholar] [CrossRef]
- Ligot, N.; Archambaud, F.; Trotta, N.; Goldman, S.; Van Bogaert, P.; Chiron, C.; De Tiège, X. Default mode network hypometabolism in epileptic encephalopathies with CSWS. Epilepsy Res. 2014, 108, 861–871. [Google Scholar] [CrossRef]
- Azeem, A.; Kirton, A.; Appendino, J.P.; Kozlik, S.; Mineyko, A. Automated quantification of spike-wave activity may be used to predict the development of electrical status epilepticus in sleep (ESES) in children with perinatal stroke. Clin. Neurophysiol. 2021, 132, 146–153. [Google Scholar] [CrossRef]
- Brandhoff, J.; de Vries, L.S.; Smal, J.C. Neonatale convulsies: Denk aan eensinustrombose [Neonatal seizures: Consider sinus thrombosis]. Ned. Tijdschr. Geneeskd. 2015, 159, A8055. [Google Scholar]
- Posar, A.; Parmeggiani, A. Neuropsychological impairment in early-onset hydrocephalus and epilepsy with continuous spike-waves during slow-wave sleep: A case report and literature review. J. Pediatr. Neurosci. 2013, 8, 141–145. [Google Scholar] [CrossRef]
- Taskin, B.D.; Tanji, K.; Feldstein, N.A.; McSwiggan-Hardin, M.; Akman, C.I. Epilepsy surgery for epileptic encephalopathy as a sequela of herpes simplex encephalitis: Case report. J. Neurosurg. Pediatr. 2017, 20, 56–63. [Google Scholar] [CrossRef]
- Hu, L.-Y.; Shi, X.-Y.; Feng, C.; Wang, J.-W.; Yang, G.; Lammers, S.H.; Yang, X.F.; Ebrahimi-Fakhari, D.; Zou, L.-P. An 8-year old boy with continuous spikes and waves during slow sleep presenting with positive onconeuronal antibodies. Eur. J. Paediatr. Neurol. 2015, 19, 257–261. [Google Scholar] [CrossRef]
- Dedeoglu, Ö.; Altaş, H.; Yılmaz, D.; Gürkaş, E.; Gülleroğlu, B.; Ekşioğlu, S.; Çıtak Kurt, N. Corpus callosum thickness: A predictive factor for the first drug efficiency of self-limited epilepsy with centrotemporal spikes (selects)? Epilepsy Res. 2023, 190, 107072. [Google Scholar] [CrossRef]
- van den Munckhof, B.; de Vries, E.E.; Braun, K.P.; Boss, H.M.; Willemsen, M.A.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Serum inflammatory mediators correlate with disease activity in electrical status epilepticus in sleep (ESES) syndrome. Epilepsia 2016, 57, e45–e50. [Google Scholar] [CrossRef]
- Ayça, S.; Aksoy, H.U.; Taştan, İ.; Polat, M. Levels of melatonin in continuous spikes and waves during sleep. J. Child Neurol. 2019, 34, 309–312. [Google Scholar] [CrossRef]
- Tarcin, G.; Aksu Uzunhan, T.; Kacar, A.; Kucur, M.; Saltik, S. The relationship between epileptic seizure and melatonin in children. Epilepsy Behav. 2020, 112, 107345. [Google Scholar] [CrossRef]
- Nicita, F.; Verrotti, A.; Pruna, D.; Striano, P.; Capovilla, G.; Savasta, S.; Spartà, M.V.; Parisi, P.; Parlapiano, G.; Tarani, L.; et al. Seizures in fetal alcohol spectrum disorders: Evaluation of clinical, electroencephalographic, and neuroradiologic features in a pediatric case series. Epilepsia 2014, 55, e60–e66. [Google Scholar] [CrossRef]
- Boronat, S.; Vicente, M.; Lainez, E.; Sánchez-Montañez, A.; Vázquez, E.; Mangado, L.; Martínez-Ribot, L.; Del Campo, M. Seizures and electroencephalography findings in 61 patients with fetal alcohol spectrum disorders. Eur. J. Med. Genet. 2017, 60, 72–78. [Google Scholar] [CrossRef]
- Pavlidis, E.; Rubboli, G.; Nikanorova, M.; Kölmel, M.S.; Gardella, E. Encephalopathy with status epilepticus during sleep (ESES) induced by oxcarbazepine in idiopathic focal epilepsy in childhood. Funct. Neurol. 2015, 30, 139–141. [Google Scholar]
- Arkilo, D.; Devinsky, O.; Mudigoudar, B.; Boronat, S.; Jennesson, M.; Sassower, K.; Vaou, O.E.; Lerner, J.T.; Jeste, S.S.; Luchsinger, K.; et al. Electroencephalographic patterns during sleep in children with chromosome 15q11.2–13.1 duplications (Dup15q). Epilepsy Behav. 2016, 57, 133–136. [Google Scholar] [CrossRef]
- Valvo, G.; Novara, F.; Brovedani, P.; Ferrari, A.R.; Guerrini, R.; Zuffardi, O.; Sicca, F. 22q11.2 microduplication syndrome and epilepsy with continuous spikes and waves during sleep (CSWS). A case report and review of the literature. Epilepsy Behav. 2012, 25, 567–572. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Labalme, A.; Hirsch, E.; Arzimanoglou, A.; Genton, P.; Motte, J.; de Saint Martin, A.; Valenti, M.P.; Boulay, C.; et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: Genomic dissection makes the link with autism. Epilepsia 2012, 53, 1526–1538. [Google Scholar] [CrossRef]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Bruneau, N.; Lozovaya, N.; Labalme, A.; Boutry-Kryza, N.; Salmi, M.; Tsintsadze, T.; Addis, L.; Motte, J.; et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat. Genet. 2013, 45, 1061–1066. [Google Scholar] [CrossRef]
- Carvill, G.L.; Regan, B.M.; Yendle, S.C.; O’Roak, B.J.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef]
- Kingwell, K. Epilepsy: GRIN2A mutations identified as key genetic drivers of epilepsy-aphasia spectrum disorders. Nat. Rev. Neurol. 2013, 9, 541. [Google Scholar] [CrossRef]
- Conroy, J.; McGettigan, P.A.; McCreary, D.; Shah, N.; Collins, K.; Parry-Fielder, B.; Moran, M.; Hanrahan, D.; Deonna, T.W.; Korff, C.M.; et al. Towards the identification of a genetic basis for Landau-Kleffner syndrome. Epilepsia 2014, 55, 858–865. [Google Scholar] [CrossRef]
- Turner, S.J.; Morgan, A.T.; Perez, E.R.; Scheffer, I.E. New genes for focal epilepsies with speech and language disorders. Curr. Neurol. Neurosci. Rep. 2015, 15, 35. [Google Scholar] [CrossRef]
- Myers, K.A.; Scheffer, I.E. GRIN2A-related speech disorders and epilepsy. In GeneReviews [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Gao, K.; Tankovic, A.; Zhang, Y.; Kusumoto, H.; Zhang, J.; Chen, W.; XiangWei, W.; Shaulsky, G.H.; Hu, C.; Traynelis, S.F.; et al. A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE 2017, 12, e0170818. [Google Scholar] [CrossRef]
- Sculier, C.; Tilmant, A.S.; De Tiège, X.; Giurgea, S.; Paquier, P.; Rudolf, G.; Lesca, G.; Van Bogaert, P. Acquired epileptic opercular syndrome related to a heterozygous deleterious substitution in GRIN2A. Epileptic Disord. 2017, 19, 345–350. [Google Scholar] [CrossRef]
- Yang, X.; Qian, P.; Xu, X.; Liu, X.; Wu, X.; Zhang, Y.; Yang, Z. GRIN2A mutations in epilepsy-aphasia spectrum disorders. Brain Dev. 2018, 40, 205–210. [Google Scholar] [CrossRef]
- Qian, P.; Yang, X.; Xu, X.; Liu, X.; Zhang, Y.; Yang, Z. Study of GRIN2A mutation in epilepsy-aphasia spectrum disorders. Chin. J. Med. Genet. 2018, 35, 314–318. [Google Scholar]
- Lesca, G.; Møller, R.S.; Rudolf, G.; Hirsch, E.; Hjalgrim, H.; Szepetowski, P. Update on the genetics of the epilepsy-aphasia spectrum and role of GRIN2A mutations. Epileptic Disord. 2019, 21, S41–S47. [Google Scholar]
- Li, X.; Xie, L.-L.; Han, W.; Hong, S.-Q.; Ma, J.-N.; Wang, J.; Jiang, L. Clinical forms and GRIN2A genotype of severe end of epileptic-aphasia spectrum disorder. Front. Pediatr. 2020, 8, 574803. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Jiao, X.; Zhang, Y.; Yang, Z. Genetic etiologies in developmental and/or epileptic encephalopathy with electrical status epilepticus during sleep: Cohort study. Front. Genet. 2021, 12, 607965. [Google Scholar] [CrossRef]
- Stanley, K.; Hostyk, J.; Tran, L.; Amengual-Gual, M.; Dugan, P.; Clark, J.; Choi, H.; Tchapyjnikov, D.; Perucca, P.; Fernandes, C.; et al. Genomic analysis of “microphenotypes” in epilepsy. Am. J. Med. Genet. A 2022, 188, 138–146. [Google Scholar] [CrossRef]
- Bonardi, C.M.; Mignot, C.; Serratosa, J.M.; Giraldez, B.G.; Moretti, R.; Rudolf, G.; Reale, C.; Gellert, P.M.; Johannesen, K.M.; Lesca, G.; et al. Expanding the clinical and EEG spectrum of CNKSR2-related encephalopathy with status epilepticus during slow sleep (ESES). Clin. Neurophysiol. 2020, 131, 1030–1039. [Google Scholar] [CrossRef]
- Higa, L.A.; Wardley, J.; Wardley, C.; Singh, S.; Foster, T.; Shen, J.J. CNKSR2-related neurodevelopmental and epilepsy disorder: A cohort of 13 new families and literature review indicating a predominance of loss of function pathogenic variants. BMC Med. Genom. 2021, 14, 186. [Google Scholar] [CrossRef]
- Arican, P.; Gencpinar, P.; Olgac Dundar, N. A new cause of developmental and epileptic encephalopathy with continuous spike-and-wave during sleep: CDKL5 disorder. Neurocase 2019, 25, 59–61. [Google Scholar] [CrossRef]
- Kapoor, D.; Majethia, P.; Anand, A.; Shukla, A.; Sharma, S. Expanding the electro-clinical phenotype of CARS2 associated neuroregression. Epilepsy Behav. Rep. 2021, 16, 100485. [Google Scholar] [CrossRef]
- Lee, I.-C.; Yang, J.-J.; Li, S.-Y. A KCNQ2 E515D mutation associated with benign familial neonatal seizures and continuous spike and waves during slow-wave sleep syndrome in Taiwan. J. Formos. Med. Assoc. 2017, 116, 711–719. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Jiao, X.R.; Zhang, Y.H.; Yang, Z.X. Genotype and phenotype of children with KCNA2 gene related developmental and epileptic encephalopathy. Chin. J. Pediatr. 2020, 58, 35–40. [Google Scholar]
- Liu, W.; Cheng, M.; Zhu, Y.; Chen, Y.; Yang, Y.; Chen, H.; Niu, X.; Tian, X.; Yang, X.; Zhang, Y. DYNC1H1-related epilepsy: Genotype-phenotype correlation. Dev. Med. Child Neurol. 2023, 65, 534–543. [Google Scholar] [CrossRef]
- Russo, A.; Gobbi, G.; Pini, A.; Møller, R.S.; Rubboli, G. Encephalopathy related to status epilepticus during sleep due to a de novo KCNA1 variant in the Kv-specific Pro-Val-Pro motif: Phenotypic description and remarkable electroclinical response to ACTH. Epileptic Disord. 2020, 22, 802–806. [Google Scholar] [CrossRef]
- Miao, P.; Tang, S.; Ye, J.; Tang, J.; Wang, J.; Zheng, C.; Li, Y.; Feng, J. Differential functional changes of Nav1.2 channel causing SCN2A-related epilepsy and status epilepticus during slow sleep. Front. Neurol. 2021, 12, 653517. [Google Scholar] [CrossRef]
- Zanni, G.; Barresi, S.; Cohen, R.; Specchio, N.; Basel-Vanagaite, L.; Valente, E.M.; Shuper, A.; Vigevano, F.; Bertini, E. A novel mutation in the endosomal Na+/H+exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res. 2014, 108, 811–815. [Google Scholar] [CrossRef]
- Coorg, R.; Weisenberg, J.L. Successful treatment of electrographic status epilepticus of sleep with felbamate in a patient with SLC9A6 mutation. Pediatr. Neurol. 2015, 53, 527–531. [Google Scholar] [CrossRef]
- Mathieu, M.L.; de Bellescize, J.; Till, M.; Flurin, V.; Labalme, A.; Chatron, N.; Sanlaville, D.; Chemaly, N.; des Portes, V.; Ostrowsky, K.; et al. Electrical status epilepticus in sleep, a constitutive feature of Christianson syndrome? Eur. J. Paediatr. Neurol. 2018, 22, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Yamamoto, A.; Ichikawa, K.; Tsuyusaki, Y.; Tsuji, M.; Iai, M.; Enomoto, Y.; Murakami, H.; Kurosawa, K.; Miyatake, S.; et al. Epilepsy in Christianson syndrome: Two cases of Lennox-Gastaut syndrome and a review of literature. Epilepsy Behav. Rep. 2019, 13, 100349. [Google Scholar] [CrossRef]
- Bhat, S.; Ming, X.; Dekermenjian, R.; Chokroverty, S. Continuous spike and wave in slow-wave sleep in a patient with Rett syndrome and in a patient with Lhermitte-Duclos syndrome and neurofibromatosis 1. J. Child Neurol. 2014, 29, NP176–NP180. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, E.; Bellini, M.; Aspromonte, M.C.; Polli, R.; Mercante, A.; Ciaccio, C.; Granocchio, E.; Bettella, E.; Donati, I.; Cainelli, E.; et al. A novel WAC loss of function mutation in an individual presenting with encephalopathy related to status epilepticus during sleep (ESES). Genes 2020, 11, 344. [Google Scholar] [CrossRef]
- Ergun-Longmire, B.; Nguyen, M.H.N.; Com, G. Electrical status epilepticus during sleep in a child with Prader-Willi syndrome: A case report. AME Case Rep. 2022, 6, 7. [Google Scholar] [CrossRef]
- Khan, A.Q.; Coorg, R.K.; Gill, D.; Marini, C.; Myers, K.A. Koolen-de Vries syndrome associated with continuous spike-wave in sleep. Epileptic Disord. 2022, 24, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, P.; Negrin, S.; Volzone, A.; Zanotta, N.; Epifanio, R.; Zucca, C.; Osanni, E.; Petacchi, E.; Fabbro, F. Electrical status epilepticus during sleep in Mowat-Wilson syndrome. Brain Dev. 2017, 39, 727–734. [Google Scholar] [CrossRef]
- Christoforou, S.; Christodoulou, K.; Anastasiadou, V.; Nicolaides, P. Early-onset presentation of a new subtype of β-Propeller protein-associated neurodegeneration (BPAN) caused by a de novo WDR45 deletion in a 6 year-old female patient. Eur. J. Med. Genet. 2020, 63, 103765. [Google Scholar] [CrossRef]
- Sager, S.G.; Turkyilmaz, A.; Gunbey, H.P.; Karatoprak, E.Y.; Aslan, E.S.; Akın, Y. A novel de novo TET3 loss-of-function variant in a Turkish boy presenting with neurodevelopmental delay and electrical status epilepticus during slow-wave sleep. Brain Dev. 2023, 45, 140–145. [Google Scholar] [CrossRef]
- Mir, A.; Amer, F.; Ali, M.; Alotaibi, W.; Alotaibi, M.; Hedaithy, A.; Aldurayhim, F.; Hussain, F.; Bashir, S.; Housawi, Y. Continuous spikes and waves during sleep (CSWS), severe epileptic encephalopathy, and choreoathetosis due to mutations in FRRS1L. Clin. EEG Neurosci. 2023, 54, 526–533. [Google Scholar] [CrossRef]
- Hadi, D.A.; Mohamed, A.R.; Rethanavelu, K.; Khoo, T.B. Clonic seizures, continuous spikes-and-waves during slow sleep, choreoathetosis and response to sulthiame in a child with FRRS1L encephalopathy. Brain Dev. 2022, 44, 44–49. [Google Scholar] [CrossRef]
- Ünalp, A.; Gazeteci Tekin, H.; Karaoğlu, P.; Akışın, Z. Benefits of ketogenic diet in a pediatric patient with Ehlers-Danlos syndrome and STXBP1-related epileptic encephalopathy. Int. J. Neurosci. 2022, 132, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, I.; Loddenkemper, T.; Peters, J.M.; Kothare, S.V. Electrical status epilepticus in sleep: Clinical presentation and pathophysiology. Pediatr. Neurol. 2012, 47, 390–410. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Conroy, J.; Deonna, T.; McCreary, D.; McGettigan, P.; Madigan, C.; Carter, I.; Ennis, S.; Lynch, S.A.; Shahwan, A.; et al. Atypical benign partial epilepsy of childhood with acquired neurocognitive, lexical semantic, and autistic spectrum disorder. Epilepsy Behav. Case Rep. 2016, 6, 42–48. [Google Scholar] [CrossRef]
- Pal, D.K.; Ferrie, C.; Addis, L.; Akiyama, T.; Capovilla, G.; Caraballo, R.; de Saint-Martin, A.; Fejerman, N.; Guerrini, R.; Hamandi, K.; et al. Idiopathic focal epilepsies: The “lost tribe”. Epileptic Disord. 2016, 18, 252–288. [Google Scholar] [CrossRef]
- Lee, Y.J.; Hwang, S.K.; Kwon, S. The clinical spectrum of benign epilepsy with centro-temporal spikes: A challenge in categorization and predictability. J. Epilepsy Res. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dryżałowski, P.; Jóźwiak, S.; Franckiewicz, M.; Strzelecka, J. Benign epilepsy with centrotemporal spikes—Current concepts of diagnosis and treatment. Neurol. Neurochir. Pol. 2018, 52, 677–689. [Google Scholar] [CrossRef]
- Halász, P.; Kelemen, A.; Rosdy, B.; Rásonyi, G.; Clemens, B.; Szűcs, A. Perisylvian epileptic network revisited. Seizure 2019, 65, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Fortini, S.; Espeche, A.; Galicchio, S.; Cersósimo, R.; Chacon, S.; Gallo, A.; Gamboni, B.; Adi, J.; Fasulo, L.; Semprino, M.; et al. More than one self-limited epilepsy of childhood in the same patient: A multicenter study. Epilepsy Res. 2021, 177, 106768. [Google Scholar] [CrossRef] [PubMed]
- Sable, S.; Sable, R.; Tamhankar, P.; Tamhankar, V. Clinical profile of patients with rolandic epilepsy at a clinic in rural Maharashtra. J. Fam. Med. Prim. Care 2021, 10, 1263–1266. [Google Scholar] [CrossRef]
- Vercueil, L. GRIN2A, a green semaphore on the lumping route to idiopathic focal epilepsy in childhood. Rev. Neurol. 2013, 169, 921–922. [Google Scholar] [CrossRef]
- Rudolf, G.; de Bellescize, J.; de Saint Martin, A.; Arzimanoglou, A.; Valenti Hirsch, M.P.; Labalme, A.; Boulay, C.; Simonet, T.; Boland, A.; Deleuze, J.F.; et al. Exome sequencing in 57 patients with self-limited focal epilepsies of childhood with typical or atypical presentations suggests novel candidate genes. Eur. J. Paediatr. Neurol. 2020, 27, 104–110. [Google Scholar] [CrossRef]
- Hausman-Kedem, M.; Menascu, S.; Greenstein, Y.; Fattal-Valevski, A. Immunotherapy for GRIN2A and GRIN2D-related epileptic encephalopathy. Epilepsy Res. 2020, 163, 106325. [Google Scholar] [CrossRef]
- Samanta, D. GRIN2A-related epilepsy and speech disorders: A comprehensive overview with a focus on the role of precision therapeutics. Epilepsy Res. 2023, 189, 107065. [Google Scholar] [CrossRef] [PubMed]
- Bebek, N.; Gürses, C.; Baykan, B.; Gökyiğit, A. Lack of prominent cognitive regression in the long-term outcome of patients having electrical status epilepticus during sleep with different types of epilepsy syndromes. Clin. EEG Neurosci. 2015, 46, 235–242. [Google Scholar] [CrossRef]
- Imataka, G.; Arisaka, O. Serial EEG study in a girl with Landau-Kleffner syndrome associated with continuous spikes and waves during slow sleep. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2145–2147. [Google Scholar]
- Posar, A.; Visconti, P. Some considerations about the association between autism spectrum disorder and epilepsy. Turk. Pediatri Ars. 2020, 55, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.-R.; Stafstrom, C.E. Pediatric epileptic encephalopathies: Pathophysiology and animal models. Semin. Pediatr. Neurol. 2016, 23, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Halász, P. Comment on neuronal networks in epileptic encephalopathies with CSWS. Epilepsia 2017, 58, 1296–1297. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Clinics | EEG | |
|---|---|---|
| Typical ESES | Cognitive deterioration. Behavioral disorders, including attention deficit, hyperactivity, aggressiveness, difficulty in social interaction, and (more rarely) psychosis; autistic behavior is also possible. Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology. Motor signs, including dyspraxia, ataxia, and dystonia. | Electrical-status epilepticus lasting for ≥85% of NREM (slow) sleep, documented by more than 2 EEG recordings during a period of ≥1 month. |
| Atypical ESES | Great heterogeneity in the reported clinical features depending on the frequency and localization of the EEG paroxysmal abnormalities: a cognitive deterioration has been reported, but less frequently than typical ESES. Behavioral disorders, in particular ADHD-like symptoms. Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology. | Paroxysmal abnormalities lasting for <85% of NREM sleep (usually > 50% < 85%), with a localization more heterogeneous than typical forms: focal, multifocal, unilateral, asymmetric or symmetric bilateral, and diffuse. |
| Clinical | Early onset of seizures; multiple types of seizures; appearance of new seizures with an increased frequency; seizure semiology including dysarthria or somatosensory auras. |
| EEG | Fronto-centro-temporal focus, with increasing frequency, both in wakefulness and in sleep; pattern EEG of spike–waves. |
| Semiautomatic quantification of paroxysmal abnormalities in CSWS is a reliable alternative to the classic quantification based on visual scoring. |
| Global synchronization increase from wakefulness to sleep is strongly correlated with spikes. |
| Associating time-sensitive magnetic source imaging and PET, spike–wave onset is associated with focal hypermetabolism. |
| Magnetoencephalography in non-lesional CSWS children showed dipole clusters located on heterogeneous cortical areas: right Rolandic area, right supramarginal gyrus, left Rolandic area, left supramarginal gyrus, bilateral Rolandic area, and multiple anatomical areas. |
| PET showed hypermetabolism in perisylvian, superior temporal, inferior parietal, and central cortex areas that were related to paroxysmal abnormalities. The diffuse hypometabolism found in regions belonging to the DMN (see prefrontal and posterior cingulate cortices, parahippocampal gyrus and precuneus) could be due to remote inhibition following epileptic activity. |
| EEG-fMRI showed characteristic findings of epileptic encephalopathy: positive changes in BOLD signals in the perisylvian regions, prefrontal cortex, anterior cingulate, and thalamus, while negative changes in BOLD signals were found in the DMN regions. The activation pattern represents a diffusion of epileptic activity. |
| HFOs are hypothesized to be related to alterations in higher brain functions. They seem to be related to the functional disruption of brain networks in CSWS. |
| Pharmacological | Antiepileptic drugs: sulthiame (++), levetiracetam (+), acetazolamide (+), benzodiazepines (++), topiramate (++), perampanel (+), lacosamide (+). Steroids (corticosteroids and ACTH) (+++). Immunoglobulins (+). Amantadine (+). |
| Non-pharmacological | Ketogenic diet (+). |
| Comments on the literature concerning these topics: almost only retrospective studies were performed, with small or very small samples of patients; there is a lack of information about the long-term effects of the drugs. Often cognitive and behavioral findings reported do not derive from standardized objective neuropsychological assessments. | |
| Unfavorable | Long duration of CSWS. Presence of a cerebral lesion. High frequency of EEG paroxysmal abnormalities. Early-onset CSWS (before 6 years). |
| Favorable | Clinical phenotype of an atypical SeLECTS at onset. Normal development before CSWS. Later-onset CSWS (from 8 years onwards). Normal EEG background organization. |
| Clinical | Acquired mixed aphasia, with onset at 3–5 years of age, with poor decoding of verbal and/or non-verbal sounds. Seizures are present in around two-thirds of cases and are semiologically heterogeneous: focal motor, tonic–clonic seizures, and atypical absences. Behavioral symptoms are frequently associated: ADHD symptoms; irritability; aggressive behavior; in some cases, autistic-like symptoms; anxiety; and depression. Learning disorders are the rule. Outcome is very heterogeneous. Language recovery completes in only a minority of cases. |
| EEG | Spikes and spike–waves prevailing in the posterior temporal regions, bilaterally, much more diffuse and frequent during non-REM sleep, becoming continuous or subcontinuous. Background activity during wakefulness and sleep is normal. |
| Pharmacological | Antiepileptic drugs: valproate (+), benzodiazepines (+), levetiracetam (+), ethosuximide (+), acetazolamide (+). Corticosteroids (+++). Immunoglobulins in cases with GRIN2A mutations (++). |
| Non-pharmacological | Ketogenic diet (+). Multiple subpial transections in selected cases (+). Vagus nerve stimulation (+). Speech therapy (++). |
| Brain structural alterations | Polymicrogyria (in particular, unilateral), migration disorders, perinatal hypoxic–ischemic encephalopathy, hydrocephalus, schizencephaly, porencephalic lesions, encephalitis, intracranial hemorrhage, and thalamic lesions. |
| Inflammation | Altered levels of interleukin-6. |
| Acquired factors | Fetal alcohol syndrome. Iatrogenic (in particular, old antiepileptic drugs). |
| Genetics | Dup15q (copy number gains of 15q11–q13), 22q11.2 microduplication, and many other copy number variants, affecting several other chromosomes, including 3q25 deletion, 3q29 duplication, 8p23.3 deletion, 10q21.3 deletion, 11p13 duplication, 16p13 deletion, and Xp22.12 deletion. Mutations of the GRIN2A gene (16p13.2). Mutations of a lot of other genes: CNKSR2 (Xp22.12), CDKL5 (Xp22.13), CARS2 (13q34), KCNQ2 (20q13.33), KCNA2 (1p13.3), SLC9A6 (Xq26.3), HIVEP2 (6q24.2), RARS2 (6q15), DYNC1H1 (14q32.31), FRRS1L (9q31.3), KCNA1 (12p13.32), SLC9A6 (Xq26.3), STXBP1 (9q34.11), MECP2 (Xq28), WAC (10p12.1), KANSL1 (17q21.31), TET3 (2p13), ZEB2 (2q22.3), and WDR45 (Xp11.23). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posar, A.; Visconti, P. Continuous Spike–Waves during Slow Sleep Today: An Update. Children 2024, 11, 169. https://doi.org/10.3390/children11020169
Posar A, Visconti P. Continuous Spike–Waves during Slow Sleep Today: An Update. Children. 2024; 11(2):169. https://doi.org/10.3390/children11020169
Chicago/Turabian StylePosar, Annio, and Paola Visconti. 2024. "Continuous Spike–Waves during Slow Sleep Today: An Update" Children 11, no. 2: 169. https://doi.org/10.3390/children11020169
APA StylePosar, A., & Visconti, P. (2024). Continuous Spike–Waves during Slow Sleep Today: An Update. Children, 11(2), 169. https://doi.org/10.3390/children11020169