
Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Diagnostics
3.1. Clinical Features
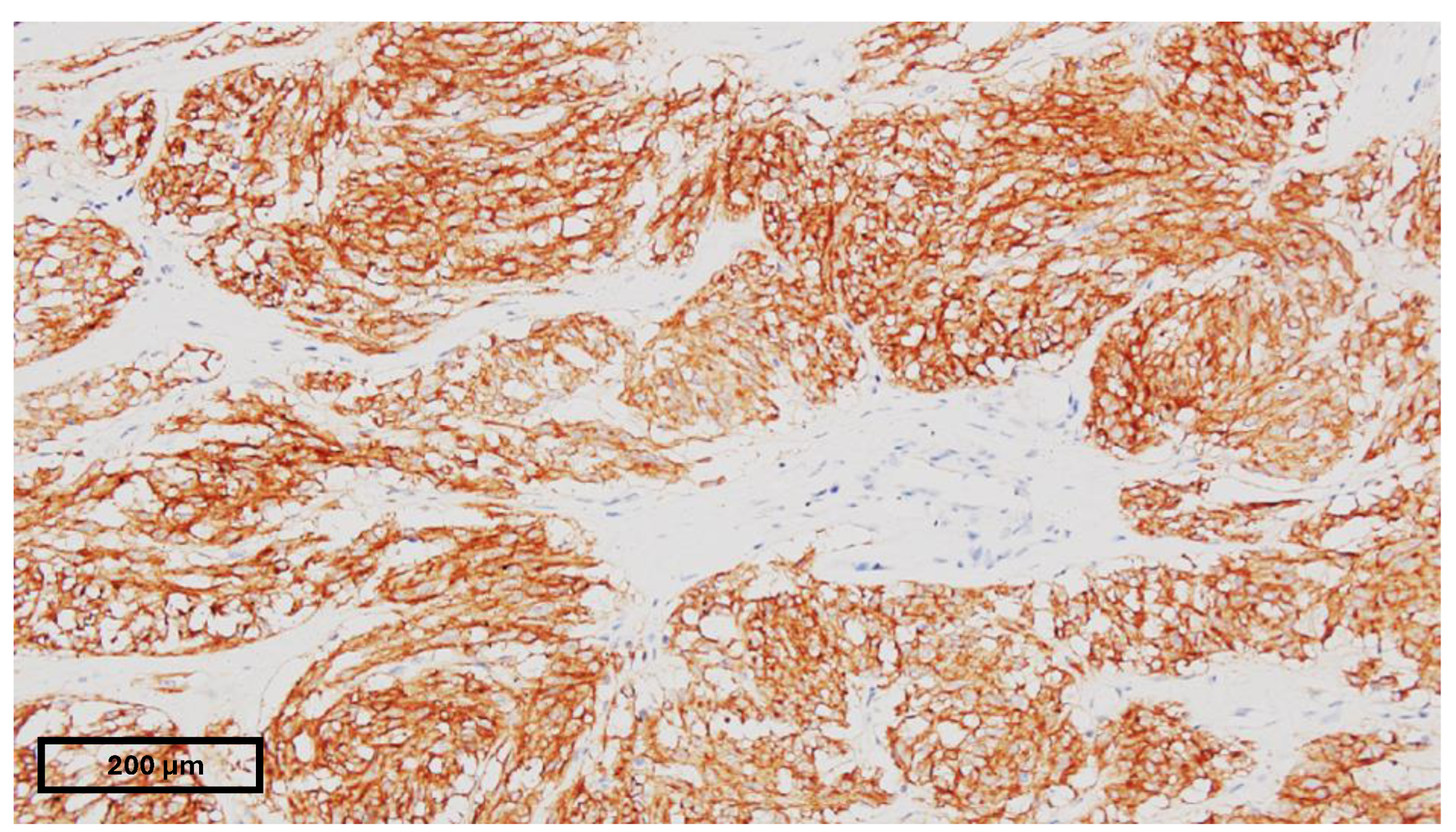
3.2. Pathology
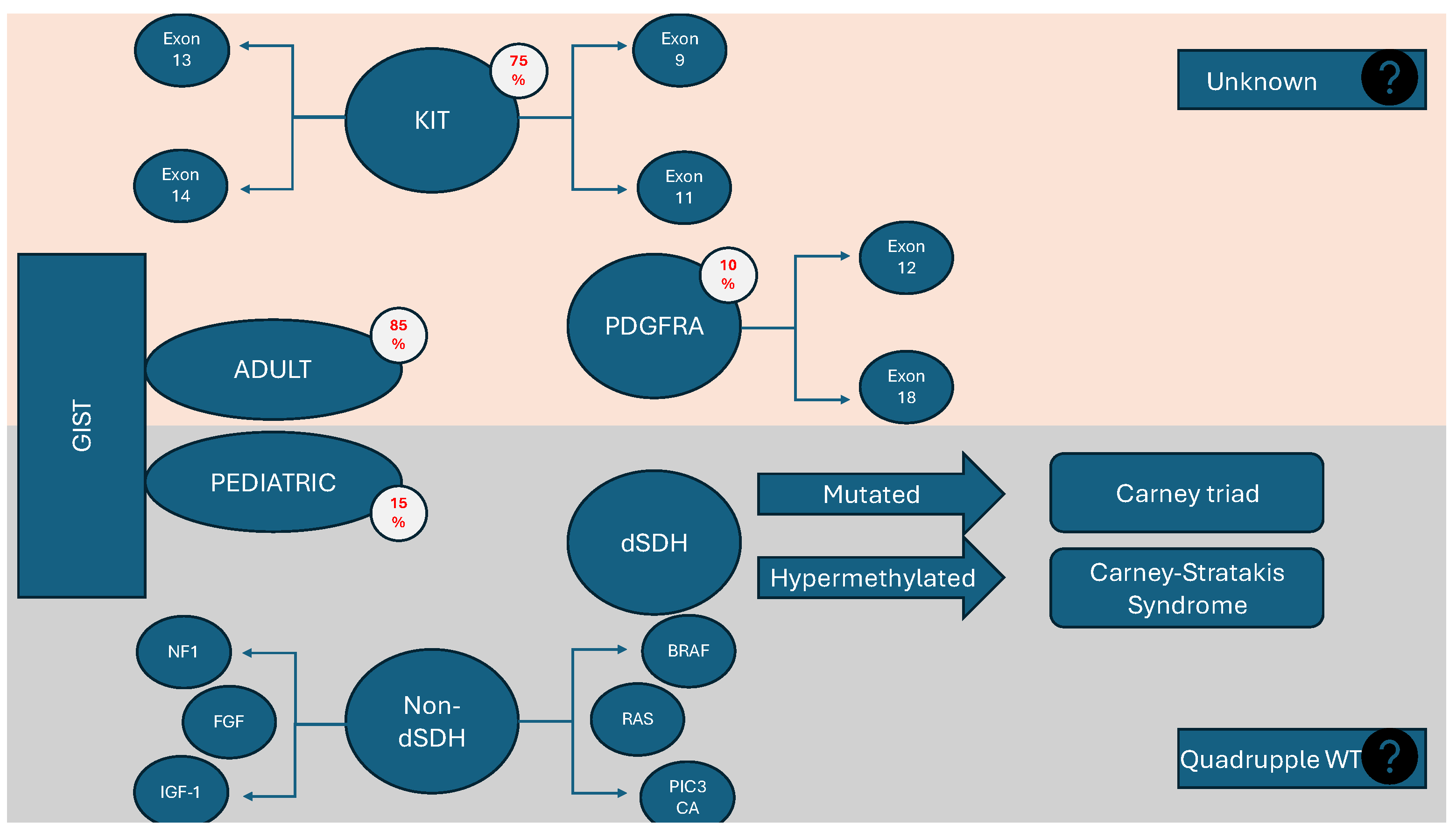
3.3. Genetics and Genotyping
3.3.1. Succinate Dehydrogenase-Deficient GISTs (dSDH)
3.3.2. BRAF and RAS Mutations
3.4. Imaging
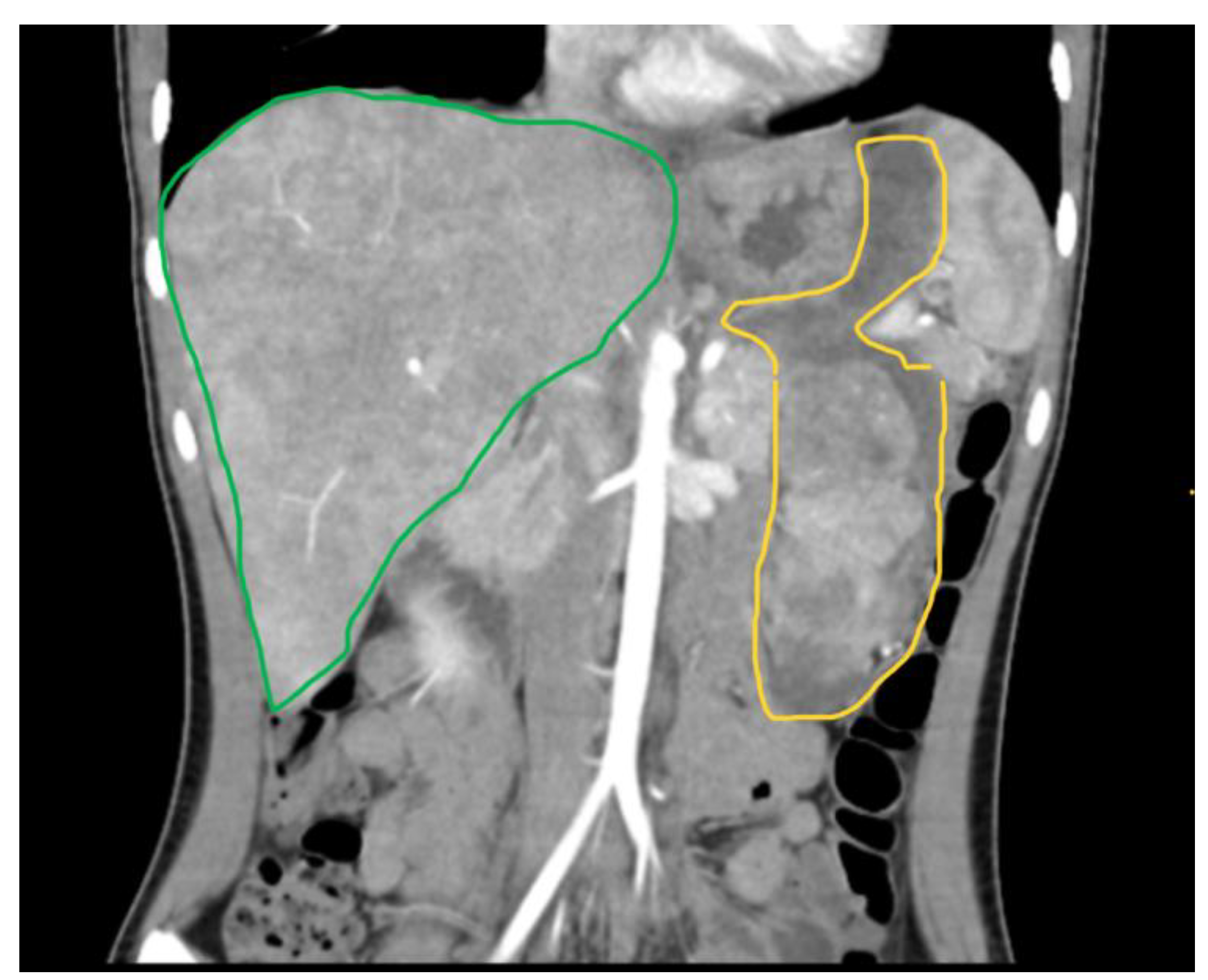
3.4.1. Computed Tomography
3.4.2. Magnetic Resonance Imaging
3.4.3. Contrast-Enhanced and Endoscopic Contrast-Enhance Ultrasound
3.5. Endoscopy
3.6. Biopsy
4. Associations with Other Pathological Entities
4.1. Carney Triad and Carney Syndrome
4.2. Neurofibromatosis Type I
5. Risk Stratification
6. Treatment
6.1. Medical Treatment
6.2. Surgical Treatment
Minimally Invasive Techniques
7. Follow-Up and Survival
8. Case Report
Case Discussion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch. Pathol. Lab. Med. 2006, 130, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Dudzisz-śledź, M.; Klimczak, A.; Bylina, E.; Rutkowski, P. Treatment of Gastrointestinal Stromal Tumors (GISTs): A Focus on Younger Patients. Cancers 2022, 14, 2831. [Google Scholar] [CrossRef]
- Min, K.W.; Leabu, M. Interstitial cells of Cajal (ICC) and gastrointestinal stromal tumor (GIST): Facts, speculations, and myths. J. Cell. Mol. Med. 2006, 10, 995–1013. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Wang, Z.F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate dehydrogenase-deficient GISTs: A clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef]
- Serrano, C.; Martín-Broto, J.; Asencio-Pascual, J.M. 2023 GEIS Guidelines for gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2023, 15, 17588359231192388. [Google Scholar] [CrossRef]
- Andrzejewska, M.; Czarny, J.; Derwich, K. Latest Advances in the Management of Pediatric Gastrointestinal Stromal Tumors. Cancers 2022, 14, 4989. [Google Scholar] [CrossRef] [PubMed]
- DeMatteo, R.P.; Lewis, J.J.; Leung, D.; Mudan, S.S.; Woodruff, J.M.; Brennan, M.F. Two hundred gastrointestinal stromal tumors: Recurrence patterns and prognostic factors for survival. Ann. Surg. 2000, 231, 51–58. [Google Scholar] [CrossRef]
- Quiroz, H.J.; Willobee, B.A.; Sussman, M.S.; Fox, B.R.; Thorson, C.M.; Sola, J.E.; Perez, E.A. Pediatric gastrointestinal stromal tumors—A review of diagnostic modalities. Transl. Gastroenterol. Hepatol. 2018, 3, 54. [Google Scholar] [CrossRef]
- Raitio, A.; Salim, A.; Mullassery, D.; Losty, P.D. Current treatment and outcomes of pediatric gastrointestinal stromal tumors (GIST): A systematic review of published studies. Pediatr. Surg. Int. 2021, 37, 1161–1165. [Google Scholar] [CrossRef]
- Theiss, L.; Contreras, C.M. Gastrointestinal Stromal Tumors of the Stomach and Esophagus. Surg. Clin. N. Am. 2019, 99, 543–553. [Google Scholar] [CrossRef]
- Kaemmer, D.A.; Otto, J.; Lassay, L.; Steinau, G.; Klink, C.; Junge, K.; Klinge, U.; Schumpelick, V. The gist of literature on pediatric GIST: Review of clinical presentation. J. Pediatr. Hematol. Oncol. 2009, 31, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rink, L.; Godwin, A.K. Clinical and molecular characteristics of gastrointestinal stromal tumors in the pediatric and young adult population. Curr. Oncol. Rep. 2009, 11, 314–321. [Google Scholar] [CrossRef]
- Tran, S.; Dingeldein, M.; Mengshol, S.C.; Kay, S.; Chin, A.C. Incidental GIST after appendectomy in a pediatric patient: A first instance and review of pediatric patients with CD117 confirmed GISTs. Pediatr. Surg. Int. 2014, 30, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Meng, X.; Gao, Y.; Yu, J.; Liu, B. A 4-month-old boy with gastrointestinal stromal tumor of mesocolon. Cancer Biol. Ther. 2019, 20, 8–14. [Google Scholar] [CrossRef]
- Hashizume, N.; Sakamoto, S.; Fukahori, S.; Ishii, S.; Saikusa, N.; Koga, Y.; Higashidate, N.; Tsuruhisa, S.; Nakahara, H.; Tanaka, Y.; et al. Gastrointestinal stromal tumor in perforated Meckel’s diverticulum: A case report and literature review. Surg. Case Rep. 2020, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Ullah, A.; Waheed, A.; Karki, N.R.; Adhikari, N.; Vemavarapu, L.; Belakhlef, S.; Bendjemil, S.M.; Mehdizadeh Seraj, S.; Sidhwa, F.; et al. Gastrointestinal Stromal Tumors (GIST): A Population-Based Study Using the SEER Database, including Management and Recent Advances in Targeted Therapy. Cancers 2022, 14, 3689. [Google Scholar] [CrossRef]
- Gold, J.S.; van der Zwan, S.M.; Gönen, M.; Maki, R.G.; Singer, S.; Brennan, M.F.; Antonescu, C.R.; De Matteo, R.P. Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Ann. Surg. Oncol. 2007, 14, 134–142. [Google Scholar] [CrossRef]
- Agaram, N.P.; Laquaglia, M.P.; Ustun, B.; Guo, T.; Wong, G.C.; Socci, N.D.; Maki, R.G.; DeMatteo, R.P.; Besmer, P.; Antonescu, C.R. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3204–3215. [Google Scholar] [CrossRef]
- Janeway, K.A.; Pappo, A. Treatment guidelines for gastrointestinal stromal tumors in children and young adults. J. Pediatr. Hematol. Oncol. 2012, 34 (Suppl. S2), S69–S72. [Google Scholar] [CrossRef]
- Zhao, X.; Yue, C. Gastrointestinal stromal tumor. J. Gastrointest. Oncol. 2012, 3, 189–208. [Google Scholar] [CrossRef]
- Nishida, T.; Blay, J.Y.; Hirota, S.; Kitagawa, Y.; Kang, Y.K. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer 2016, 19, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Demoulin, S.; Trenquier, E.; Dekkar, S.; Deshayes, S.; Boisguérin, P.; Serrano, C.; de Santa Barbara, P.; Faure, S. LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib. Int. J. Mol. Sci. 2023, 24, 7138. [Google Scholar] [CrossRef] [PubMed]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Urbini, M.; Astolfi, A.; Indio, V.; Nannini, M.; Schipani, A.; Bacalini, M.G.; Angelini, S.; Ravegnini, G.; Calice, G.; Del Gaudio, M.; et al. Gene duplication, rather than epigenetic changes, drives FGF4 overexpression in KIT/PDGFRA/SDH/RAS-P WT GIST. Sci. Rep. 2020, 10, 19829. [Google Scholar] [CrossRef]
- Steeghs, E.M.P.; Kroeze, L.I.; Tops, B.B.J.; van Kempen, L.C.; Ter Elst, A.; Kastner-van Raaij, A.W.M.; Hendriks-Cornelissen, S.J.B.; Hermsen, M.J.W.; Jansen, E.A.M.; Nederlof, P.M.; et al. Comprehensive routine diagnostic screening to identify predictive mutations, gene amplifications, and microsatellite instability in FFPE tumor material. BMC Cancer 2020, 20, 291. [Google Scholar] [CrossRef]
- Atiq, M.A.; Davis, J.L.; Hornick, J.L.; Dickson, B.C.; Fletcher, C.D.M.; Fletcher, J.A.; Folpe, A.L.; Mariño-Enríquez, A. Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements: A clinicopathological, immunophenotypic, and molecular study of eight cases, emphasizing their distinction from gastrointestinal stromal tumor (GIST). Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2021, 34, 95–103. [Google Scholar] [CrossRef]
- Huss, S.; Pasternack, H.; Ihle, M.A.; Merkelbach-Bruse, S.; Heitkötter, B.; Hartmann, W.; Trautmann, M.; Gevensleben, H.; Büttner, R.; Schildhaus, H.-U.; et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum. Pathol. 2017, 62, 206–214. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Ibrahim, A.; Chopra, S. Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Arch. Pathol. Lab. Med. 2020, 144, 655–660. [Google Scholar] [CrossRef]
- Giger, O.T.; Ten Hoopen, R.; Shorthouse, D.; Abdullahi, S.; Bulusu, V.R.; Jadhav, S.; Maher, E.R.; Casey, R.T. Preferential MGMT hypermethylation in SDH-deficient wild-type GIST. J. Clin. Pathol. 2023, 77, 34–39. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.-F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, F.; Guo, Q.H.; Wang, Y.P.; Zhao, R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J. Gastroenterol. 2020, 26, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Huang, C.-J.; Wu, M.-T.; Yang, S.-F.; Su, Y.-C.; Chai, C.-Y. Hypoxia-inducible factor-1alpha is associated with risk of aggressive behavior and tumor angiogenesis in gastrointestinal stromal tumor. Jpn. J. Clin. Oncol. 2005, 35, 207–213. [Google Scholar] [CrossRef]
- Bai, C.; Liu, X.; Qiu, C.; Zheng, J. FoxM1 is regulated by both HIF-1α and HIF-2α and contributes to gastrointestinal stromal tumor progression. Gastric Cancer 2019, 22, 91–103. [Google Scholar] [CrossRef]
- Kalfusova, A.; Linke, Z.; Kalinova, M.; Krskova, L.; Hilska, I.; Szabova, J.; Vicha, A.; Kodet, R. Gastrointestinal stromal tumors—Summary of mutational status of the primary/secondary KIT/PDGFRA mutations, BRAF mutations and SDH defects. Pathol. Res. Pract. 2019, 215, 152708. [Google Scholar] [CrossRef]
- Jašek, K.; Váňová, B.; Grendár, M.; Štanclová, A.; Szépe, P.; Hornáková, A.; Holubeková, V.; Plank, L.; Lasabová, Z. BRAF mutations in KIT/PDGFRA positive gastrointestinal stromal tumours (GISTs): Is their frequency underestimated? Pathol. Res. Pract. 2020, 216, 153171. [Google Scholar] [CrossRef]
- Niinuma, T.; Suzuki, H.; Sugai, T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl. Gastroenterol. Hepatol. 2018, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Nishida, T.; Naito, Y.; Takahashi, T.; Saito, T.; Hisamori, S.; Manaka, D.; Ogawa, K.; Hirota, S.; Ichikawa, H. Molecular and clinicopathological features of KIT/PDGFRA wild-type gastrointestinal stromal tumors. Cancer Sci. 2024, 115, 894–904. [Google Scholar] [CrossRef]
- Franck, C.; Rosania, R.; Franke, S.; Haybaeck, J.; Canbay, A.; Venerito, M. The BRAF Status May Predict Response to Sorafenib in Gastrointestinal Stromal Tumors Resistant to Imatinib, Sunitinib, and Regorafenib: Case Series and Review of the Literature. Digestion 2019, 99, 179–184. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Kalkmann, J.; Zeile, M.; Antoch, G.; Berger, F.; Diederich, S.; Dinter, D.; Fink, C.; Janka, R.; Stattaus, J. Consensus report on the radiological management of patients with gastrointestinal stromal tumours (GIST): Recommendations of the German GIST Imaging Working Group. Cancer Imaging 2012, 12, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Bano, S.; Puri, S.K.; Upreti, L.; Chaudhary, V.; Sant, H.K.; Gondal, R. Gastrointestinal stromal tumors (GISTs): An imaging perspective. Jpn. J. Radiol. 2012, 30, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Tsurumaru, D.; Nishimuta, Y.; Kai, S.; Oki, E.; Minoda, Y.; Ishigami, K. Clinical significance of dual-energy dual-layer CT parameters in differentiating small-sized gastrointestinal stromal tumors from leiomyomas. Jpn. J. Radiol. 2023, 41, 1389–1396. [Google Scholar] [CrossRef]
- Janeway, K.A.; Albritton, K.H.; Van Den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr. Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, X.; Zeng, Y.; Wu, R.; Ding, L.; Xia, Y.; Chen, Z.; Zhang, X.; Wang, X. [18F]FAPI-42 PET/CT versus [18107F]FDG PET/CT for imaging of recurrent or metastatic gastrointestinal stromal tumors. Eur. J. Nucl. Med. Mol. Imaging 2022, 50, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, M.; Beer, M.; Anupindi, S.; Vollert, K.; Kröncke, T. Imaging pediatric gastrointestinal stromal tumor (GIST). J. Pediatr. Surg. 2018, 53, 1862–1870. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, D.; Zheng, T.; Liu, D.; Fang, Y. Predicting the progression-free survival of gastrointestinal stromal tumors after imatinib therapy through multi-sequence magnetic resonance imaging. Abdom. Radiol. 2024, 49, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.H.; Lee, J.M.; Baek, J.H.; Han, J.K.; Choi, B.-I. MRI features of gastrointestinal stromal tumors. AJR Am. J. Roentgenol. 2014, 203, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Charles-Edwards, E.M.; De Souza, N.M. Diffusion-weighted magnetic resonance imaging and its application to cancer. Cancer Imaging 2006, 6, 135–143. [Google Scholar] [CrossRef]
- Dietrich, C.; Hartung, E.; Ignee, A. The use of contrast-enhanced ultrasound in patients with GIST metastases that are negative in CT and PET. Ultraschall Med. 2008, 29 (Suppl. S5), 276–277. [Google Scholar] [CrossRef]
- Ignee, A.; Jenssen, C.; Hocke, M.; Dong, Y.; Wang, W.-P.; Cui, X.-W.; Woenckhaus, M.; Iordache, S.; Saftoiu, A.; Schuessler, G.; et al. Contrast-enhanced (endoscopic) ultrasound and endoscopic ultrasound elastography in gastrointestinal stromal tumors. Endosc. Ultrasound 2017, 6, 55–60. [Google Scholar] [CrossRef]
- Okasha, H.H.; Naguib, M.; El Nady, M.; Ezzat, R.; Al-Gemeie, E.; Al-Nabawy, W.; Aref, W.; Abdel-Moaty, A.; Essam, K.; Hamdy, A. Role of endoscopic ultrasound and endoscopic-ultrasound-guided fine-needle aspiration in endoscopic biopsy negative gastrointestinal lesions. Endosc. Ultrasound 2017, 6, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Chhoda, A.; Jain, D.; Surabhi, V.R.; Singhal, S. Contrast enhanced harmonic endoscopic ultrasound: A novel approach for diagnosis and management of gastrointestinal stromal tumors. Clin. Endosc. 2018, 51, 215–221. [Google Scholar] [CrossRef]
- Wu, J.; Zhuang, M.; Zhou, Y.; Zhan, X.; Xie, W. The value of contrast-enhanced harmonic endoscopic ultrasound in differential diagnosis and evaluation of malignant risk of gastrointestinal stromal tumors (<50 mm). Scand. J. Gastroenterol. 2023, 58, 542–548. [Google Scholar] [CrossRef]
- Facciorusso, A.; Crinò, S.F.; Ramai, D.; Ofosu, A.; Muscatiello, N.; Mangiavillano, B.; Lamonaca, L.; Lisotti, A.; Fusaroli, P.; Gkolfakis, P.; et al. Comparison between endoscopic ultrasound-guided fine-needle biopsy and bite-on-bite jumbo biopsy for sampling of subepithelial lesions. Dig. liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2022, 54, 676–683. [Google Scholar] [CrossRef]
- Jacobson, B.C.; Bhatt, A.; Greer, K.B.; Lee, L.S.; Park, W.G.; Sauer, B.G.; Shami, V.M. ACG Clinical Guideline: Diagnosis and Management of Gastrointestinal Subepithelial Lesions. Am. J. Gastroenterol. 2023, 118, 46–58. [Google Scholar] [CrossRef]
- Jakob, J.; Salameh, R.; Wichmann, D.; Charalambous, N.; Zygmunt, A.C.; Kreisel, I.; Heinz, J.; Ghadimi, M.; Ronellenfitsch, U. Needle tract seeding and abdominal recurrence following pre-treatment biopsy of gastrointestinal stromal tumors (GIST): Results of a systematic review. BMC Surg. 2022, 22, 202. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Rulyak, S.D.; Kimmey, M.B. American Gastroenterological Association Institute Technical Review on the Management of Gastric Subepithelial Masses. Gastroenterology 2006, 130, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef]
- Pitsava, G.; Settas, N.; Faucz, F.R.; Stratakis, C.A. Carney Triad, Carney-Stratakis Syndrome, 3PAS and Other Tumors Due to SDH Deficiency. Front. Endocrinol. 2021, 12, 680609. [Google Scholar] [CrossRef]
- Killian, J.K.; Miettinen, M.; Walker, R.L.; Wang, Y.; Zhu, Y.J.; Waterfall, J.J.; Noyes, N.; Retnakumar, P.; Yang, Z.; Smith, W.I.J.; et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci. Transl. Med. 2014, 6, 268ra177. [Google Scholar] [CrossRef]
- Matyakhina, L.; Bei, T.A.; McWhinney, S.R.; Pasini, B.; Cameron, S.; Gunawan, B.; Stergiopoulos, S.G.; Boikos, S.; Muchow, M.; Dutra, A.; et al. Genetics of carney triad: Recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2938–2943. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A.; Stratakis, C.A. Familial paraganglioma and gastric stromal sarcoma: A new syndrome distinct from the Carney triad. Am. J. Med. Genet. 2002, 108, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.-Q.; Jiang, Y.-P.; Yi, B.-H.; Yang, Y.; Sun, D.-Z.; Fan, J.-X. Neurofibromatosis type 1 with multiple gastrointestinal stromal tumors: A case report. World J. Clin. Cases 2023, 11, 2336–2342. [Google Scholar] [CrossRef]
- Takazawa, Y.; Sakurai, S.; Sakuma, Y.; Ikeda, T.; Yamaguchi, J.; Hashizume, Y.; Yokoyama, S.; Motegi, A.; Fukayama, M. Gastrointestinal stromal tumors of neurofibromatosis type I (von Recklinghausen’s disease). Am. J. Surg. Pathol. 2005, 29, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-negative GIST is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef]
- Hudgi, A.R.; Azam, M.; Masood, M.; Arshad, H.M.S.; Yap, J.E.L. The Gastrointestinal Stromal Tumors of It: A Rare Presentation of Neurofibromatosis Type I. Cureus 2021, 13, e16034. [Google Scholar] [CrossRef]
- Joensuu, H.; Vehtari, A.; Riihimäki, J.; Nishida, T.; Steigen, S.E.; Brabec, P.; Plank, L.; Nilsson, B.; Cirilli, C.; Braconi, C.; et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: An analysis of pooled population-based cohorts. Lancet. Oncol. 2012, 13, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum. Pathol. 2002, 33, 459–465. [Google Scholar] [CrossRef]
- Gold, J.S.; Gönen, M.; Gutiérrez, A.; Broto, J.M.; García-del-Muro, X.; Smyrk, T.C.; Maki, R.G.; Singer, S.; Brennan, M.F.; Antonescu, C.R.; et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: A retrospective analysis. Lancet Oncol. 2009, 10, 1045–1052. [Google Scholar] [CrossRef]
- Joensuu, H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum. Pathol. 2008, 39, 1411–1419. [Google Scholar] [CrossRef]
- Hemming, M.L.; Coy, S.; Lin, J.-R.; Andersen, J.L.; Przybyl, J.; Mazzola, E.; Abdelhamid Ahmed, A.H.; van de Rijn, M.; Sorger, P.K.; Armstrong, S.A.; et al. HAND1 and BARX1 Act as Transcriptional and Anatomic Determinants of Malignancy in Gastrointestinal Stromal Tumor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 1706–1719. [Google Scholar] [CrossRef]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Schlemmer, M.; Bauer, S.; Schütte, R.; Hartmann, J.T.; Bokemeyer, C.; Hosius, C.; Reichardt, P. Activity and side effects of imatinib in patients with gastrointestinal stromal tumors: Data from a German multicenter trial. Eur. J. Med. Res. 2011, 16, 206–212. [Google Scholar] [CrossRef]
- Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The emerging role of the FGF/FGFR pathway in gastrointestinal stromal tumor. Int. J. Mol. Sci. 2020, 21, 3313. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Glod, J.; Arnaldez, F.I.; Wiener, L.; Spencer, M.; Killian, J.K.; Meltzer, P.; Dombi, E.; Derse-Anthony, C.; Derdak, J.; Srinivasan, R.; et al. A Phase II Trial of Vandetanib in Children and Adults with Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2019, 25, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Rusakiewicz, S.; Perier, A.; Semeraro, M.; Pitt, J.M.; von Strandmann, E.P.; Reiners, K.S.; Aspeslagh, S.; Pipéroglou, C.; Vély, F.; Ivagnes, A.; et al. NKp30 isoforms and NKp30 ligands are predictive biomarkers of response to imatinib mesylate in metastatic GIST patients. Oncoimmunology 2017, 6, e1137418. [Google Scholar] [CrossRef]
- Casali, P.G.; Blay, J.Y.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; et al. Gastrointestinal stromal tumours: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up ☆. Ann. Oncol. 2022, 33, 20–33. [Google Scholar] [CrossRef]
- Reichardt, P. The Story of Imatinib in GIST—A Journey through the Development of a Targeted Therapy. Oncol. Res. Treat. 2018, 41, 472–477. [Google Scholar] [CrossRef]
- Trent, J.C.; Subramanian, M.P. Managing GIST in the imatinib era: Optimization of adjuvant therapy. Expert Rev. Anticancer Ther. 2014, 14, 1445–1459. [Google Scholar] [CrossRef]
- Rihacek, M.; Selingerova, I.; Kocak, I.; Kocakova, I.; Rihackova, E.; Valik, D.; Sterba, J. Sunitinib-Induced Elevation of Mean Corpuscular Volume (MCV)—Exploring Its Possible Clinical Relevance in Cancer Patients. Curr. Oncol. 2022, 29, 4138–4147. [Google Scholar] [CrossRef]
- Prior, J.O.; Montemurro, M.; Orcurto, M.-V.; Michielin, O.; Luthi, F.; Benhattar, J.; Guillou, L.; Elsig, V.; Stupp, R.; Delaloye, A.B.; et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 439–445. [Google Scholar] [CrossRef]
- Verschuur, A.C.; Bajčiová, V.; Mascarenhas, L.; Khosravan, R.; Lin, X.; Ingrosso, A.; Janeway, K.A. Sunitinib in pediatric patients with advanced gastrointestinal stromal tumor: Results from a phase I/II trial. Cancer Chemother. Pharmacol. 2019, 84, 41–50. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Daudigeos-Dubus, E.; Le Dret, L.; Lanvers-Kaminsky, C.; Bawa, O.; Opolon, P.; Vievard, A.; Villa, I.; Pagès, M.; Bosq, J.; Vassal, G.; et al. Regorafenib: Antitumor Activity upon Mono and Combination Therapy in Preclinical Pediatric Malignancy Models. PLoS ONE 2015, 10, e0142612. [Google Scholar] [CrossRef]
- Brinch, C.; Dehnfeld, M.; Hogdall, E.; Poulsen, T.S.; Toxvaerd, A.; Al-Farra, G.; Bergenfeldt, M.; Krarup-Hansen, A. Outstanding Response to Sorafenib in a Patient with Metastatic Gastrointestinal Stromal Tumour. Case Rep. Oncol. 2021, 14, 1567–1573. [Google Scholar] [CrossRef]
- George, S.; von Mehren, M.; Fletcher, J.A.; Sun, J.; Zhang, S.; Pritchard, J.R.; Hodgson, J.G.; Kerstein, D.; Rivera, V.M.; Haluska, F.G.; et al. Phase II Study of Ponatinib in Advanced Gastrointestinal Stromal Tumors: Efficacy, Safety, and Impact of Liquid Biopsy and Other Biomarkers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1268–1276. [Google Scholar] [CrossRef]
- Garner, A.P.; Gozgit, J.M.; Anjum, R.; Vodala, S.; Schrock, A.; Zhou, T.; Serrano, C.; Eilers, G.; Zhu, M.; Ketzer, J.; et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5745–5755. [Google Scholar] [CrossRef]
- Trent, J.C.; Wathen, K.; von Mehren, M.; Samuels, B.L.; Staddon, A.P.; Choy, E.; Butrynski, J.E.; Chugh, R.; Chow, W.A.; Rushing, D.A.; et al. A phase II study of dasatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST). J. Clin. Oncol. 2011, 29, 10006. [Google Scholar] [CrossRef]
- van Tilburg, C.M.; DuBois, S.G.; Albert, C.M.; Federman, N.; Nagasubramanian, R.; Geoerger, B.; Orbach, D.; Bielack, S.S.; Shukla, N.N.; Turpin, B.; et al. Larotrectinib efficacy and safety in pediatric TRK fusion cancer patients. J. Clin. Oncol. 2019, 37, 10010. [Google Scholar] [CrossRef]
- Kang, Y.-K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef]
- Serrano, C.; Bauer, S.; Gómez-Peregrina, D.; Kang, Y.-K.; Jones, R.L.; Rutkowski, P.; Mir, O.; Heinrich, M.C.; Tap, W.D.; Newberry, K.; et al. Circulating tumor DNA analysis of the phase III VOYAGER trial: KIT mutational landscape and outcomes in patients with advanced gastrointestinal stromal tumor treated with avapritinib or regorafenib. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 615–625. [Google Scholar] [CrossRef]
- Ligon, J.A.; Sundby, R.T.; Wedekind, M.F.; Arnaldez, F.I.; Del Rivero, J.; Wiener, L.; Srinivasan, R.; Spencer, M.; Carbonell, A.; Lei, H.; et al. A Phase II Trial of Guadecitabine in Children and Adults with SDH-Deficient GIST, Pheochromocytoma, Paraganglioma, and HLRCC-Associated Renal Cell Carcinoma. Clin. Cancer Res. 2022, 29, 341–348. [Google Scholar] [CrossRef]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef]
- Von Mehren, M.; George, S.; Heinrich, M.C.; Schuetze, S.M.; Yap, J.T.; Yu, J.Q.; Abbott, A.; Litwin, S.; Crowley, J.; Belinsky, M.; et al. Linsitinib (OSI-906) for the treatment of adult and pediatric wild-type gastrointestinal stromal tumors, a SARC phase II study. Clin. Cancer Res. 2020, 26, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, J.; Nakajima, K.; Omori, T.; Takahashi, T.; Nishitani, A.; Ito, T.; Nishida, T. Surgical strategy for gastric gastrointestinal stromal tumors: Laparoscopic vs. open resection. Surg. Endosc. 2007, 21, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Weldon, C.B. Pediatric gastrointestinal stromal tumor. Semin. Pediatr. Surg. 2012, 21, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Valadão, M.; de Mello, E.L.R.; Lourenço, L.; Vilhena, B.; Romano, S.; dos Castro, L.S. What is the prognostic significance of metastatic lymph nodes in GIST? Hepatogastroenterology 2008, 55, 471–474. [Google Scholar]
- Li, C.; Su, D.; Xie, C.; Chen, Q.; Zhou, J.; Wu, X. Lymphadenectomy is associated with poor survival in patients with gastrointestinal stromal tumors. Ann. Transl. Med. 2019, 7, 558. [Google Scholar] [CrossRef]
- Stiles, Z.E.; Fleming, A.M.; Dickson, P.V.; Tsao, M.; Glazer, E.S.; Shibata, D.; Deneve, J.L. Lymph Node Metastases in Gastrointestinal Stromal Tumors: An Uncommon Event. Ann. Surg. Oncol. 2022, 29, 8641–8648. [Google Scholar] [CrossRef]
- Agaimy, A.; Wünsch, P.H. Lymph node metastasis in gastrointestinal stromal tumours (GIST) occurs preferentially in young patients < or =40 years: An overview based on our case material and the literature. Langenbeck’s Arch. Surg. 2009, 394, 375–381. [Google Scholar] [CrossRef]
- Weldon, C.B.; Madenci, A.L.; Boikos, S.A.; Janeway, K.A.; George, S.; von Mehren, M.; Pappo, A.S.; Schiffman, J.D.; Wright, J.; Trent, J.C.; et al. Surgical Management of Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Pediatric and Wildtype GIST Clinic. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 523–528. [Google Scholar] [CrossRef]
- Krajinovic, K.; Germer, C.T.; Agaimy, A.; Wünsch, P.H.; Isbert, C. Outcome after resection of one hundred gastrointestinal stromal tumors. Dig. Surg. 2010, 27, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.; Hohenberger, P. Neoadjuvant therapy to downstage the extent of resection of gastrointestinal stromal tumors. Visc. Med. 2018, 34, 359–365. [Google Scholar] [CrossRef]
- Hølmebakk, T.; Bjerkehagen, B.; Boye, K.; Bruland, Ø.; Stoldt, S.; Sundby Hall, K. Definition and clinical significance of tumour rupture in gastrointestinal stromal tumours of the small intestine. Br. J. Surg. 2016, 103, 684–691. [Google Scholar] [CrossRef]
- Petrasova, N.; Snajdauf, J.; Petru, O.; Frybova, B.; Svojgr, K.; Linke, Z.; Mixa, V.; Kodet, R.; Kyncl, M.; Rygl, M. Gastric tumors in children: Single-center study with emphasis on treatment of repeated recurrence. Pediatr. Surg. Int. 2020, 36, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Hiki, N.; Yamamoto, Y.; Fukunaga, T.; Yamaguchi, T.; Nunobe, S.; Tokunaga, M.; Miki, A.; Ohyama, S.; Seto, Y. Laparoscopic and endoscopic cooperative surgery for gastrointestinal stromal tumor dissection. Surg. Endosc. 2008, 22, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Aisu, Y.; Yasukawa, D.; Kimura, Y.; Hori, T. Laparoscopic and endoscopic cooperative surgery for gastric tumors: Perspective for actual practice and oncological benefits. World J. Gastrointest. Oncol. 2018, 10, 381–397. [Google Scholar] [CrossRef]
- Kikuchi, S.; Nishizaki, M.; Kuroda, S.; Tanabe, S.; Noma, K.; Kagawa, S.; Shirakawa, Y.; Kato, H.; Okada, H.; Fujiwara, T. Nonexposure laparoscopic and endoscopic cooperative surgery (closed laparoscopic and endoscopic cooperative surgery) for gastric submucosal tumor. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2017, 20, 553–557. [Google Scholar] [CrossRef]
- Pulido, J.; Garavito, J.; Franco, L.; Padilla, L.; Cabrera, F.; Pedraza, M.; Villarreal, R.; Bernal, F. Laparoendoscopic surgery for the treatment of gastrointestinal stromal tumors: A case series. Cir. Y Cir. (Engl. Ed.) 2022, 90 (Suppl. S1), 121–126. [Google Scholar] [CrossRef]
- Teng, T.Z.J.; Ishraq, F.; Chay, A.F.T.; Tay, K.V. Lap-Endo cooperative surgery (LECS) in gastric GIST: Updates and future advances. Surg. Endosc. 2023, 37, 1672–1682. [Google Scholar] [CrossRef]
- Onimaru, M.; Inoue, H.; Ikeda, H.; Abad, M.R.A.; Quarta Colosso, B.M.; Shimamura, Y.; Sumi, K.; Deguchi, Y.; Ito, H.; Yokoyama, N. Combination of laparoscopic and endoscopic approaches for neoplasia with non-exposure technique (CLEAN-NET) for gastric submucosal tumors: Updated advantages and limitations. Ann. Transl. Med. 2019, 7, 582. [Google Scholar] [CrossRef]
- Hiki, N.; Nunobe, S.; Matsuda, T.; Hirasawa, T.; Yamamoto, Y.; Yamaguchi, T. Laparoscopic endoscopic cooperative surgery. Dig. Endosc. 2015, 27, 197–204. [Google Scholar] [CrossRef]
- Hiki, N.; Nunobe, S. Laparoscopic endoscopic cooperative surgery (LECS) for the gastrointestinal tract: Updated indications. Ann. Gastroenterol. Surg. 2019, 3, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Fero, K.E.; Coe, T.; Fanta, P. Surgical Management of adolescents and young adults with GIST A US Population-based analysis. JAMA Surg. 2017, 152, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Yonkus, J.A.; Alva-Ruiz, R.; Grotz, T.E. Surgical Management of Metastatic Gastrointestinal Stromal Tumors. Curr. Treat. Options Oncol. 2021, 22, 37. [Google Scholar] [CrossRef]
- Zamulko, O.Y.; Zamulko, A.O.; Dawson, M.J. Introducing GIST and Dieulafoy—Think of Them in GI Bleeding and Anemia. S. D. Med. 2019, 72, 528–530. [Google Scholar] [PubMed]
- Sorour, M.A.; Kassem, M.I.; Ghazal, A.E.-H.A.; El-Riwini, M.T.; Abu Nasr, A. Gastrointestinal stromal tumors (GIST) related emergencies. Int. J. Surg. 2014, 12, 269–280. [Google Scholar] [CrossRef]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef]
- André, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with Dovitinib (TKI258): Preclinical and Clinical Data in Breast Cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef]
- Camillo Porta Palma Giglione, W.L.; Paglino, C. Dovitinib (CHIR258, TKI258): Structure, Development and Preclinical and Clinical Activity. Futur. Oncol. 2015, 11, 39–50. [Google Scholar] [CrossRef]
- Boichuk, S.; Dunaev, P.; Skripova, V.; Galembikova, A.; Bikinieva, F.; Shagimardanova, E.; Gazizova, G.; Deviatiiarov, R.; Valeeva, E.; Mikheeva, E.; et al. Unraveling the Mechanisms of Sensitivity to Anti-FGF Therapies in Imatinib-Resistant Gastrointestinal Stromal Tumors (GIST) Lacking Secondary KIT Mutations. Cancers 2023, 15, 5354. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popoiu, T.-A.; Pîrvu, C.-A.; Popoiu, C.-M.; Iacob, E.R.; Talpai, T.; Voinea, A.; Albu, R.-S.; Tãban, S.; Bãlãnoiu, L.-M.; Pantea, S. Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review. Children 2024, 11, 1040. https://doi.org/10.3390/children11091040
Popoiu T-A, Pîrvu C-A, Popoiu C-M, Iacob ER, Talpai T, Voinea A, Albu R-S, Tãban S, Bãlãnoiu L-M, Pantea S. Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review. Children. 2024; 11(9):1040. https://doi.org/10.3390/children11091040
Chicago/Turabian StylePopoiu, Tudor-Alexandru, Cãtãlin-Alexandru Pîrvu, Cãlin-Marius Popoiu, Emil Radu Iacob, Tamas Talpai, Amalia Voinea, Rãzvan-Sorin Albu, Sorina Tãban, Larisa-Mihaela Bãlãnoiu, and Stelian Pantea. 2024. "Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review" Children 11, no. 9: 1040. https://doi.org/10.3390/children11091040
APA StylePopoiu, T.-A., Pîrvu, C.-A., Popoiu, C.-M., Iacob, E. R., Talpai, T., Voinea, A., Albu, R.-S., Tãban, S., Bãlãnoiu, L.-M., & Pantea, S. (2024). Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review. Children, 11(9), 1040. https://doi.org/10.3390/children11091040