
Unique Pulmonary Hypertension in Young Children: A Case Series Study
 , , and
, , and
Abstract
:1. Introduction
- Prenatal or developmental pulmonary vascular hypertensive disease;
- Perinatal pulmonary vascular maladaptation;
- Pediatric cardiovascular disease;
- Bronchopulmonary dysplasia (BPD);
- Isolated pediatric pulmonary vascular hypertensive disease;
- Multifactorial pediatric pulmonary vascular hypertensive disease associated with other birth defects;
- Pediatric lung disease;
- Pediatric thromboembolic disease;
- Pediatric hypobaric hypoxic exposure;
- Pediatric pulmonary hypertensive vascular disease associated with other systemic disorders.
2. Materials and Methods
3. Results
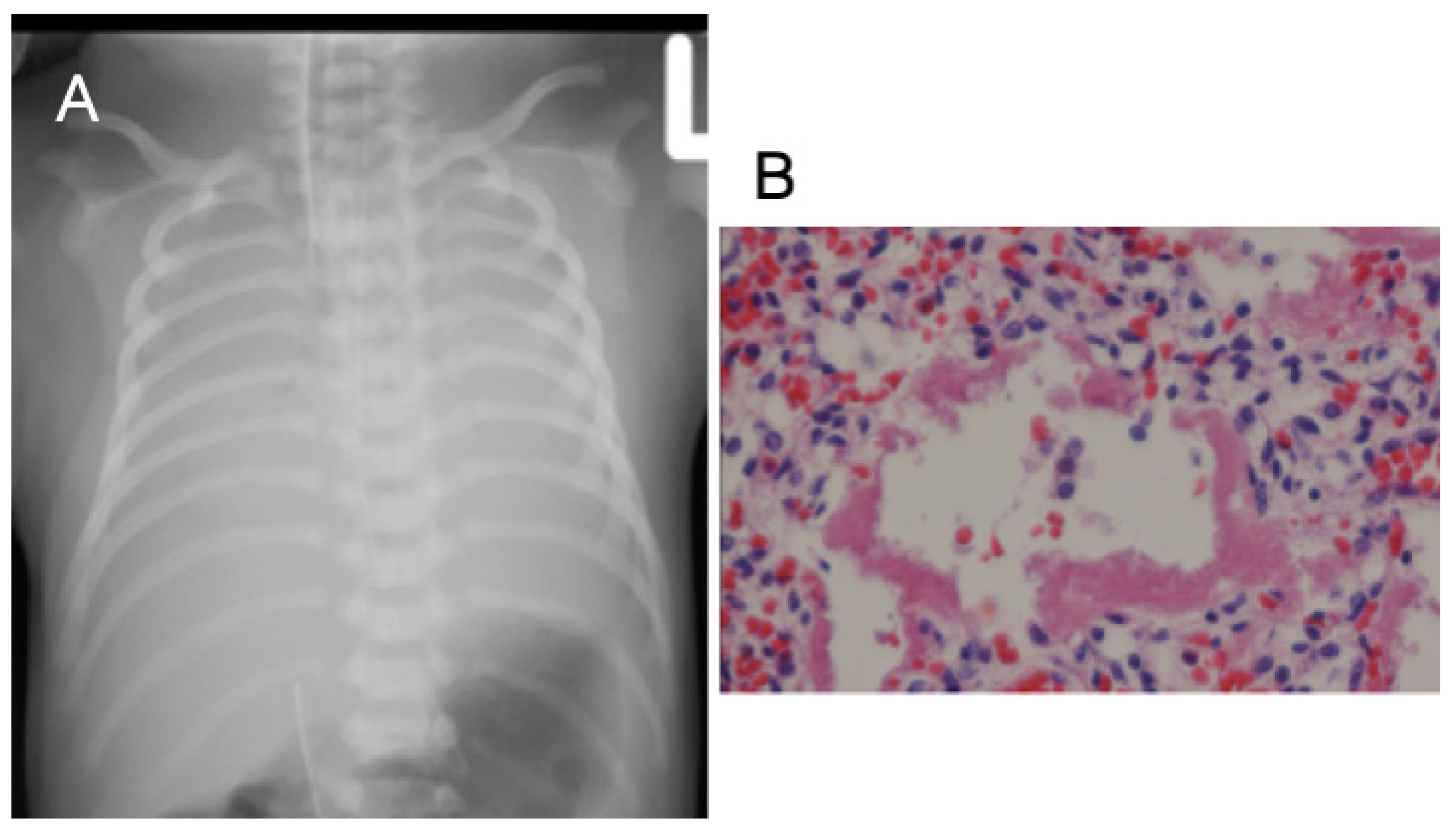
3.1. Case 1
3.2. Case 2
3.3. Case 3
3.4. Case 4
3.5. Case 5
3.6. Case 6
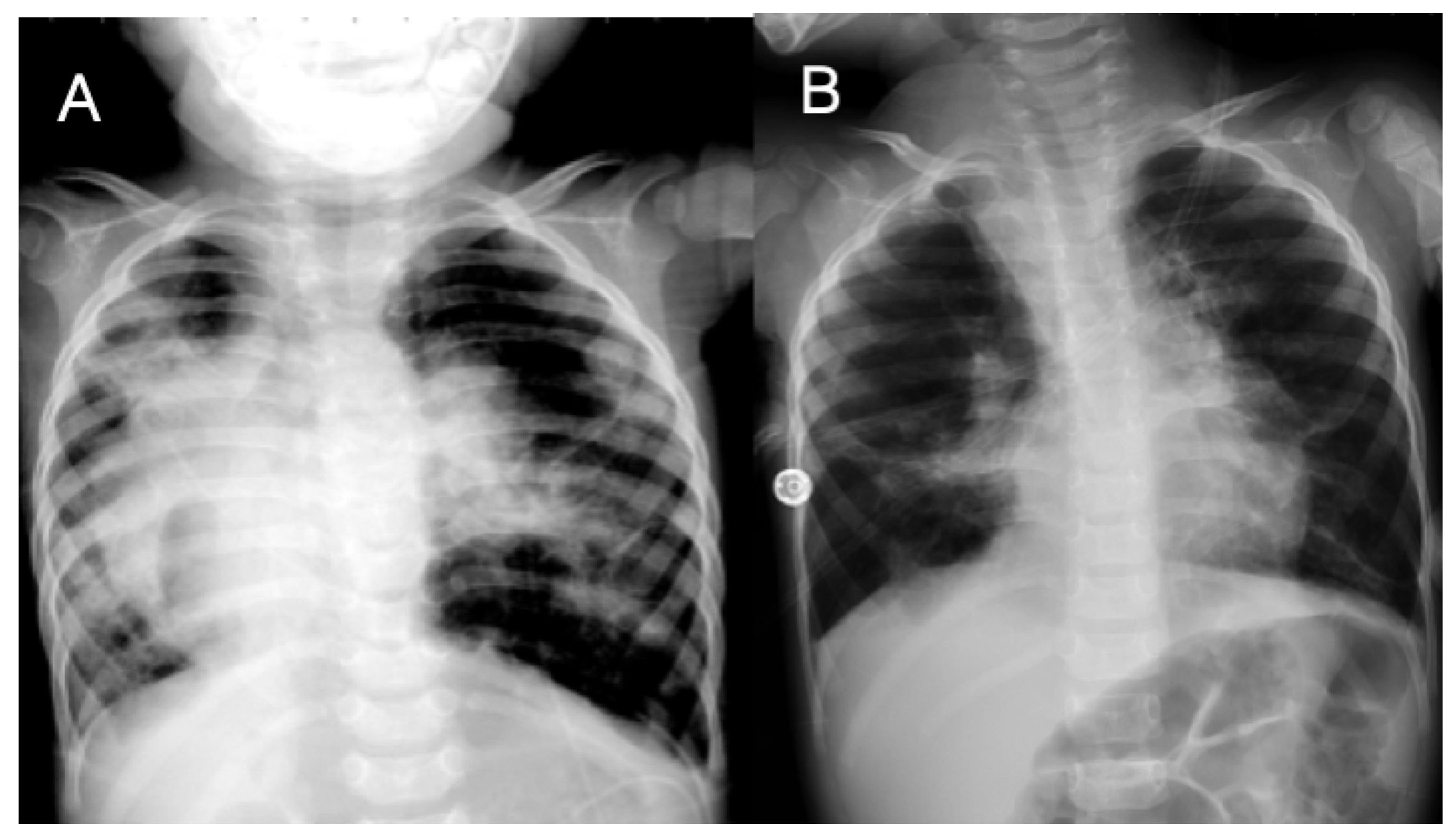
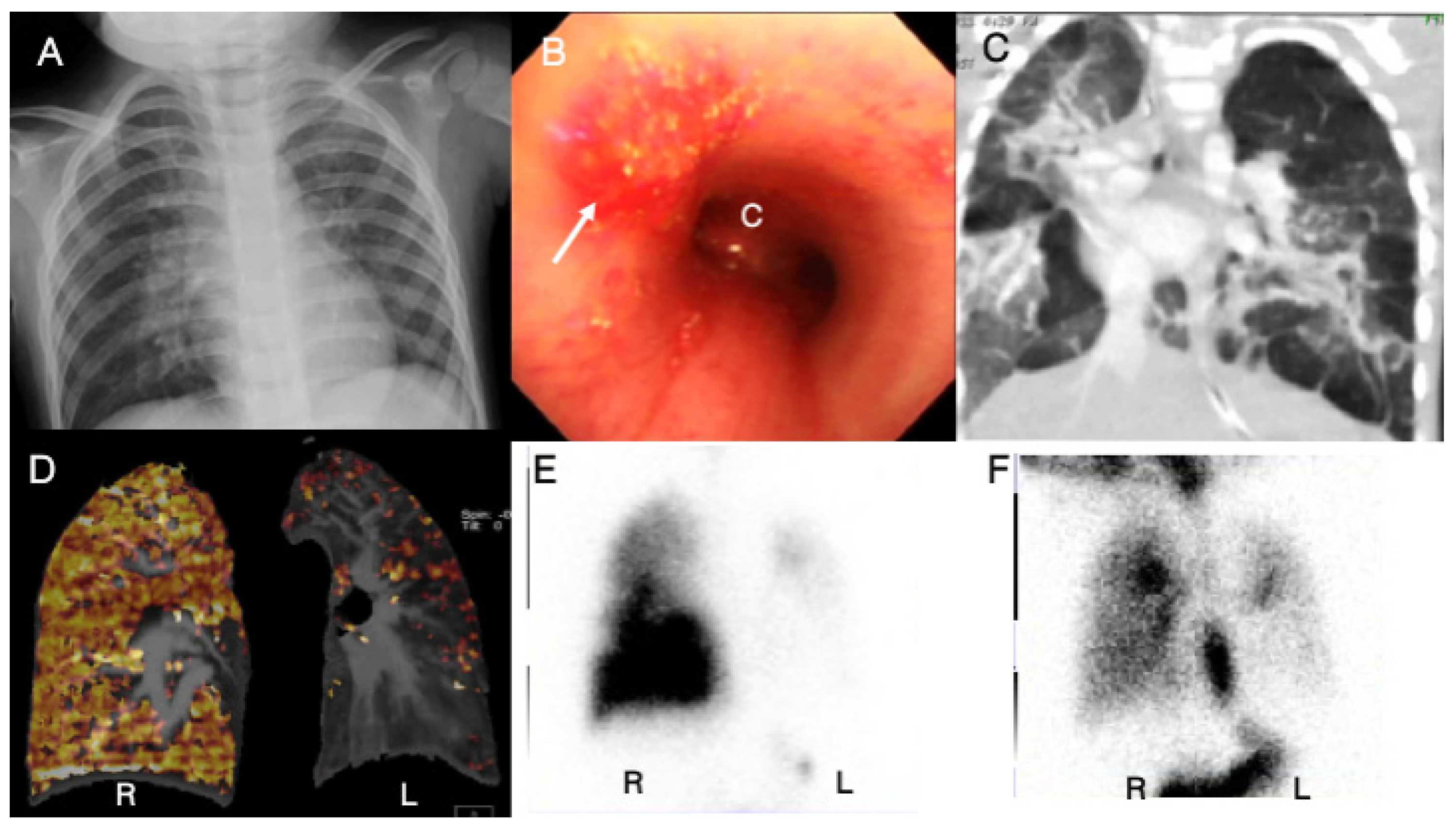
3.7. Case 7
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, G.; Bassareo, P.P.; Barilla, F.; Martino, F.; Fanos, V.; Fedele, F.; Romeo, F. Pulmonary hypertension in pediatrics. A feasible approach to bridge the gap between real world and guidelines. J. Matern. Fetal. Neonatal. Med. 2021, 34, 3820–3826. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J. Classification of pediatric pulmonary hypertensive vascular disease: Does it need to be different from the adult classification? Pulm. Circ. 2011, 1, 134–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerro, M.J.; Abman, S.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, A.E.; Adatia, I.; Cerro, M.J.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. Functional classification of pulmonary hypertension in children: Report from the PVRI pediatric taskforce, Panama 2011. Pulm. Circ. 2011, 1, 280–285. [Google Scholar] [CrossRef]
- Cueto-Robledo, G.; Jurado-Hernandez, M.Y.; Camacho-Delgado, F.R.; Roldan-Valadez, E.; Heredia-Arroyo, A.L.; Cueto-Romero, H.D.; Palafox, L.E.G.; Anaya, R.O.; Dircio, A.R.; Vazquez, H.M.; et al. Pulmonary thromboendarterectomy in Klinefelter Syndrome. Literature review. Curr. Probl. Cardiol. 2021, 101003. [Google Scholar] [CrossRef]
- Sharma, M.; Mohan, K.R.; Narayan, S.; Chauhan, L. Persistent pulmonary hypertension of the newborn: A review. Med. J. Armed. Forces India 2011, 67, 348–353. [Google Scholar] [CrossRef]
- Pathirana, J.; Munoz, F.M.; Abbing-Karahagopian, V.; Bhat, N.; Harris, T.; Kapoor, A.; Keene, D.L.; Mangili, A.; Padula, M.A.; Pande, S.L.; et al. Neonatal death: Case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 2016, 34, 6027–6037. [Google Scholar] [CrossRef] [Green Version]
- Goss, K. Long-term pulmonary vascular consequences of perinatal insults. J. Physiol. 2019, 597, 1175–1184. [Google Scholar] [CrossRef]
- Zivanovic, S.; Peacock, J.; Alcazar-Paris, M.; Lo, J.W.; Lunt, A.; Marlow, N.; Calvert, S.; Greenough, A. Late outcomes of a randomized trial of high-frequency oscillation in neonates. N. Engl. J. Med. 2014, 370, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Altit, G.; Bhombal, S.; Hopper, R.K.; Tacy, T.A.; Feinstein, J. Death or resolution: The "natural history" of pulmonary hypertension in bronchopulmonary dysplasia. J. Perinatol. 2019, 39, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghanem, G.; Shah, P.; Thomas, S.; Banfield, L.; El Helou, S.; Fusch, C.; Mukerji, A. Bronchopulmonary dysplasia and pulmonary hypertension: A meta-analysis. J. Perinatol. 2017, 37, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, J.L.; Pakrashi, T.; Jones, D.E.; South, A.P.; Shah, T.A. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J. Perinatol. 2011, 31, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Bhat, R.; Salas, A.A.; Foster, C.; Carlo, W.A.; Ambalavanan, N. Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics 2012, 129, e682–e689. [Google Scholar] [CrossRef] [Green Version]
- Arattu Thodika, F.M.S.; Nanjundappa, M.; Dassios, T.; Bell, A.; Greenough, A. Pulmonary hypertension in infants with bronchopulmonary dysplasia: Risk factors, mortality and duration of hospitalisation. J. Perinat. Med. 2022, 50, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Gorenflo, M.; Ivy, D.D.; Moledina, S.; Bonnet, D. Treatment of pediatric pulmonary arterial hypertension: A focus on the NO-sGC-cGMP pathway. Pediatr. Pulmonol. 2019, 54, 1516–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, G.; Sallmon, H.; Roehr, C.C.; Kourembanas, S.; Austin, E.D.; Koestenberger, M.; European Pediatric Pulmonary Vascular Disease Network. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr. Res. 2021, 89, 446–455. [Google Scholar] [CrossRef]
- Cohen, J.L.; Nees, S.N.; Valencia, G.A.; Rosenzweig, E.B.; Krishnan, U.S. Sildenafil use in children with pulmonary hypertension. J. Pediatr. 2019, 205, 29–34.e1. [Google Scholar] [CrossRef]
- Kadmon, G.; Schiller, O.; Dagan, T.; Bruckheimer, E.; Birk, E.; Schonfeld, T. Pulmonary hypertension specific treatment in infants with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2017, 52, 77–83. [Google Scholar] [CrossRef]
- Chen, I.C.; Lin, J.Y.; Liu, Y.C.; Chai, C.Y.; Yeh, J.L.; Hsu, J.H.; Wu, B.N.; Dai, Z.K. The beneficial effects of angiotensin-converting enzyme II (ACE2) activator in pulmonary hypertension secondary to left ventricular dysfunction. Int. J. Med. Sci. 2020, 17, 2594–2602. [Google Scholar] [CrossRef]
- Chen, I.C.; Lin, J.Y.; Liu, Y.C.; Chai, C.Y.; Yeh, J.L.; Hsu, J.H.; Wu, B.N.; Dai, Z.K. Angiotensin-Converting Enzyme 2 Activator Ameliorates Severe Pulmonary Hypertension in a Rat Model of Left Pneumonectomy Combined With VEGF Inhibition. Front. Med. 2021, 8, 619133. [Google Scholar] [CrossRef] [PubMed]
- de Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; Francois, C.; Schalij, I.; Dorfmuller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Gong, Y.; Xiao, Z.; Guang, X.; Yin, X. Decreased levels of serum angiotensin-(1-7) in patients with pulmonary arterial hypertension due to congenital heart disease. Int. J. Cardiol. 2014, 176, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Gao, L.; Wang, Q.; Chen, X.; Na, J.; Xu, X.; Yuan, Y. Angiotensinogen M235T polymorphism and susceptibility to hypertrophic cardiomyopathy in Asian population: A meta analysis. J. Renin. Angiotensin Aldosterone Syst. 2020, 21, 1470320320978100. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.E.; Jung, J.K.; Lee, Y.H.; Jang, B.C.; In Kim, J. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn Schmiedebergs Arch Pharm. 2020, 393, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Tonna-DeMasi, M.; Park, E.; Schuller-Levis, G.; Quinn, M.R. Taurine chloramine inhibits production of nitric oxide and prostaglandin E2 in activated C6 glioma cells by suppressing inducible nitric oxide synthase and cyclooxygenase-2 expression. Brain Res. Mol. Brain Res. 1998, 59, 189–195. [Google Scholar] [CrossRef]
- Jang, H.J.; Kim, S.J. Taurine exerts anti-osteoclastogenesis activity via inhibiting ROS generation, JNK phosphorylation and COX-2 expression in RAW264.7 cells. J. Recept. Signal Transduct. Res. 2013, 33, 387–391. [Google Scholar] [CrossRef]
- Dirweesh, A.; Alvarez, C.; Khan, M.; Shah, N. A unilateral hyperlucent lung-Swyer-James syndrome: A case report and literature review. Respir. Med. Case Rep. 2017, 20, 104–106. [Google Scholar] [CrossRef]
- Chen, I.C.; Chen, Y.W.; Lin, S.H.; Hsu, J.H.; Wu, J.R.; Dai, Z.K. Usefulness of combination of pulmonary ventilation and perfusion scintigraphy on the diagnosis of children with unilateral hyperlucent lung. Nucl. Med. Commun. 2011, 32, 1052–1059. [Google Scholar] [CrossRef]
- Yuce, M.; Bayram, N.; Ozer, O.; Davutoglu, V. An unusual cause of pulmonary hypertension: Swyer-James-Macleod syndrome. Int. J. Cardiol. 2016, 223, 212–214. [Google Scholar] [CrossRef]
- Capela, C.; Gouveia, P.; Sousa, M.; Regadas, M.J. Adult diagnosis of Swyer-James-MacLeod syndrome: A case report. J. Med. Case Rep. 2011, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez Fabrellas, E.; Marin Gonzalez, M.; Almenar Bonet, L.; Blanquer Olivas, R. Severe pulmonary hypertension: A feature of Swyer-James syndrome? Monaldi. Arch Chest. Dis. 1997, 52, 140–141. [Google Scholar] [PubMed]
- Sakai, M.; Kiguchi, T.; Suzuki, S.; Hitomi, H.; Sugiyama, K.; Takeda, J.; Kudoh, K.; Matsuoka, T.; Takatani, O. A case of Swyer-James syndrome diagnosed at age 70. Nihon Kyobu Shikkan Gakkai Zasshi 1990, 28, 994–998. [Google Scholar] [PubMed]
- Bode, S.F.N.; Uhl, M.; Heinzmann, A. Persistent pulmonary hypertension as clue for Swyer-James-MacLeod Syndrome. Klin. Padiatr. 2022, 234, 113–115. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. | Age/Sex | Special Consideration | Classification | SPAP (mmHg) | Major Medical Treatment to PH | Prognosis | ||
|---|---|---|---|---|---|---|---|---|
| 6th WSPH [1] | Panama [4] | Simplified [2] | ||||||
| 1 | 1 d/M | 47XXY | 1 | IV | I | 70 | PGE1 | Expired (1 d) |
| 2 | 52 d/M | Idiopathic | 1 | II | I | 84 | F + D + S + iNO + iPGI2 | Expired (3 mo) |
| 3 | 1 y/M | BPD | 3 | IV | I | 93 | F + S + iPGI2 | Expired (3 y) |
| 4 | 1 y/F | BPD | 3 | IV | I | 66 | F + S + iNO | Stable |
| 5 | 1 d/F | High renin and aldosterone | 5 | VI | I | 95 | S + F + iNO + ARB + ACEI | Expired (7 y) |
| 6 | 1 d/F | Taurine | unclear | I | I | 55 | F+iNO | Stable |
| 7 | 3 y/M | Swyer–James syndrome | 5 | V | III | 59 | F + D + S + ACEI | Expired (4 y) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, I.-C.; Chen, H.-L.; Liu, Y.-C.; Wu, Y.-H.; Lo, S.-H.; Hsu, J.-H.; Yin, H.-L.; Hsu, J.-S.; Wu, B.-N.; Dai, Z.-K. Unique Pulmonary Hypertension in Young Children: A Case Series Study. Children 2022, 9, 1064. https://doi.org/10.3390/children9071064
Chen I-C, Chen H-L, Liu Y-C, Wu Y-H, Lo S-H, Hsu J-H, Yin H-L, Hsu J-S, Wu B-N, Dai Z-K. Unique Pulmonary Hypertension in Young Children: A Case Series Study. Children. 2022; 9(7):1064. https://doi.org/10.3390/children9071064
Chicago/Turabian StyleChen, I-Chen, Hsiu-Lin Chen, Yi-Ching Liu, Yen-Hsien Wu, Shih-Hsing Lo, Jong-Hau Hsu, Hsin-Ling Yin, Jui-Sheng Hsu, Bin-Nan Wu, and Zen-Kong Dai. 2022. "Unique Pulmonary Hypertension in Young Children: A Case Series Study" Children 9, no. 7: 1064. https://doi.org/10.3390/children9071064