

Nature-Derived Compounds as Potential Bioactive Leads against CDK9-Induced Cancer: Computational and Network Pharmacology Approaches

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Ligand Preparation
2.3. Molecular Docking
2.4. Analyzing the ADMET Properties of the Selected Ligands
2.5. Molecular Dynamics
2.6. Network-Pharmacology-Based Mechanism Analyses
2.7. Construction of the Compound–Target–Pathway Network
3. Results
3.1. Protein Preparation
3.2. Ligand Preparation
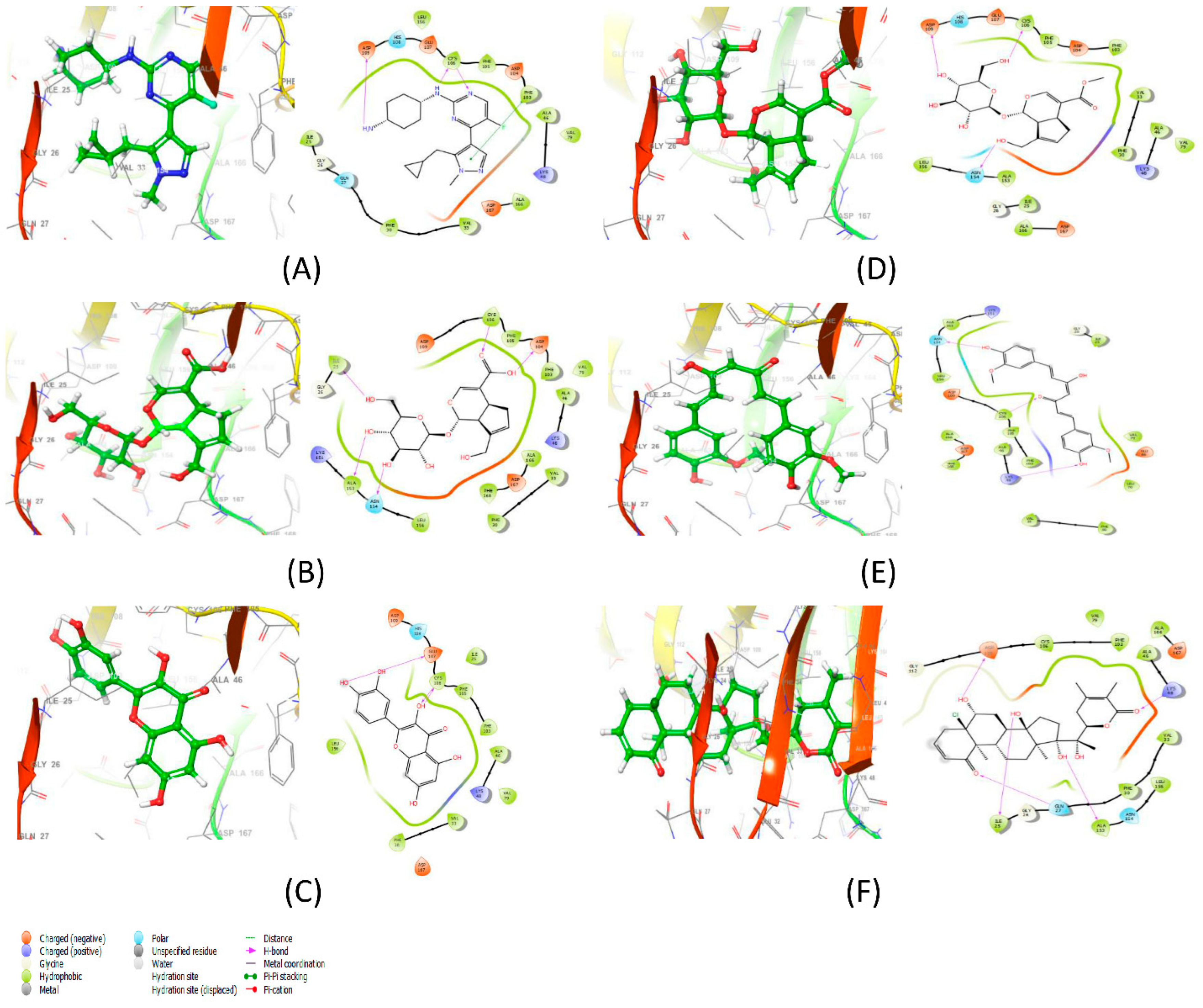
3.3. Molecular Docking Results Analysis and Binding Interaction Determination
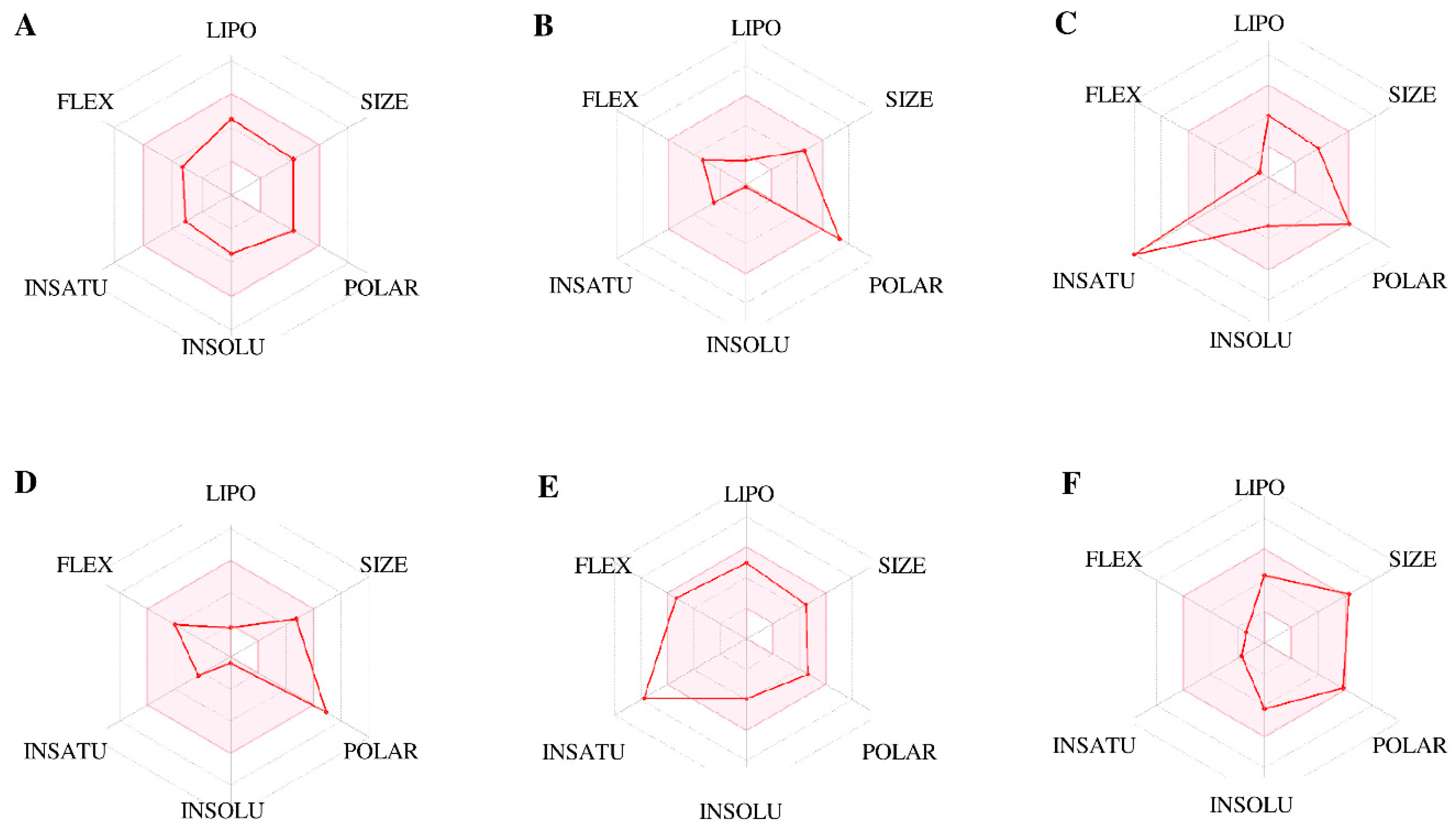
3.4. ADMET Prediction and Anticipation of the Selected Ligands
3.5. Molecular Dynamics
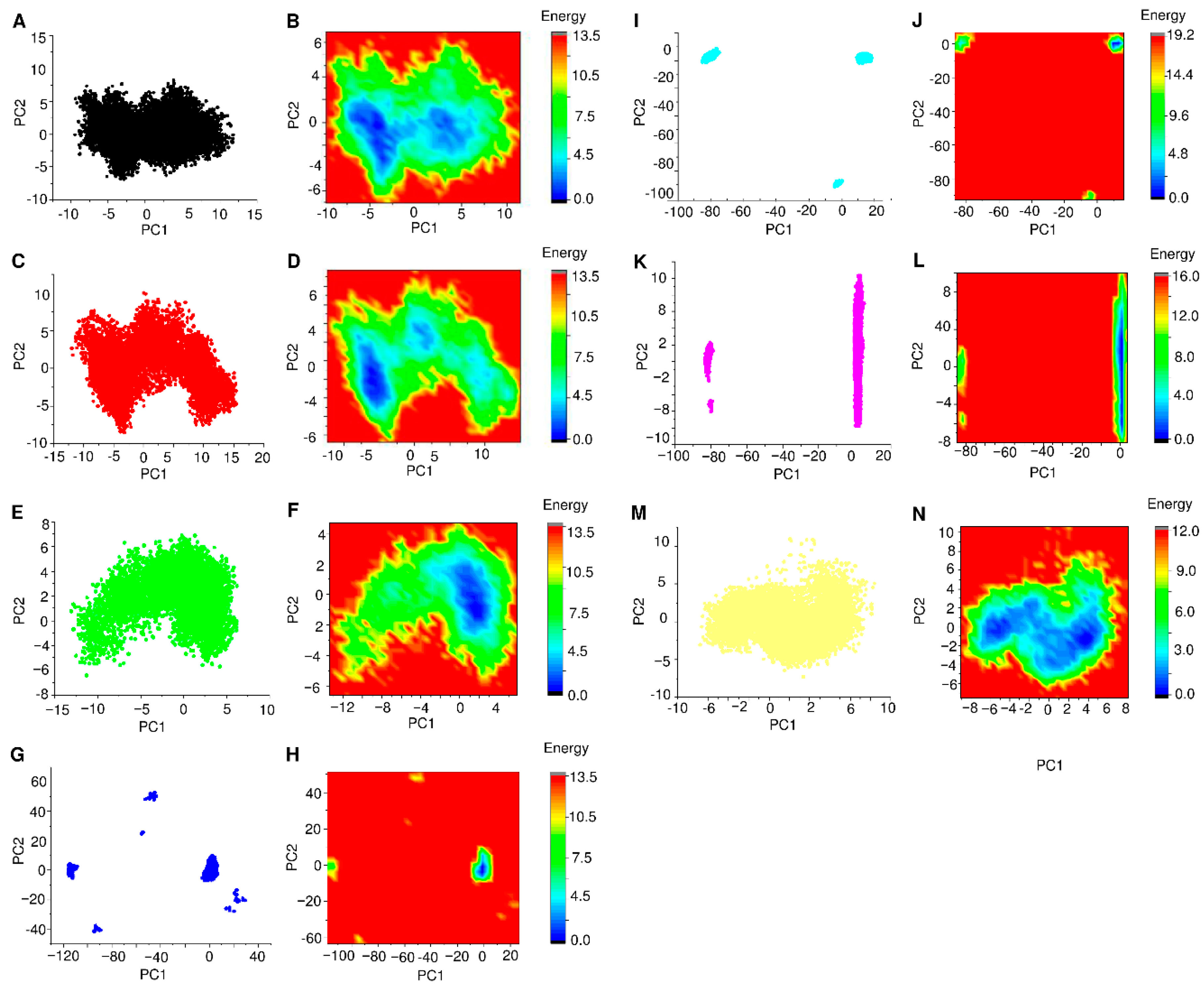
3.6. PCA and FEL Analysis
3.7. Network Pharmacology Analysis
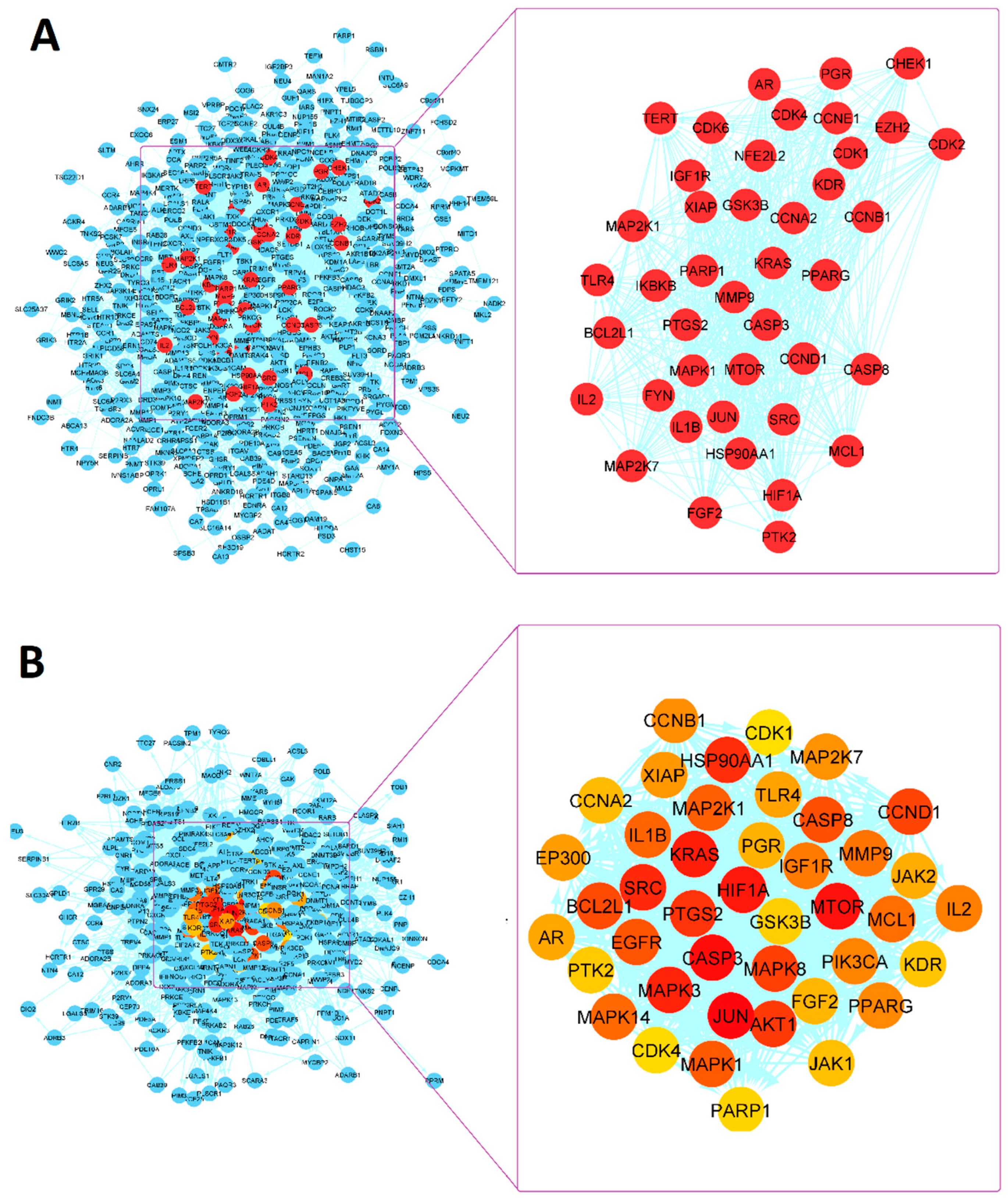
3.7.1. PPI Network Analysis
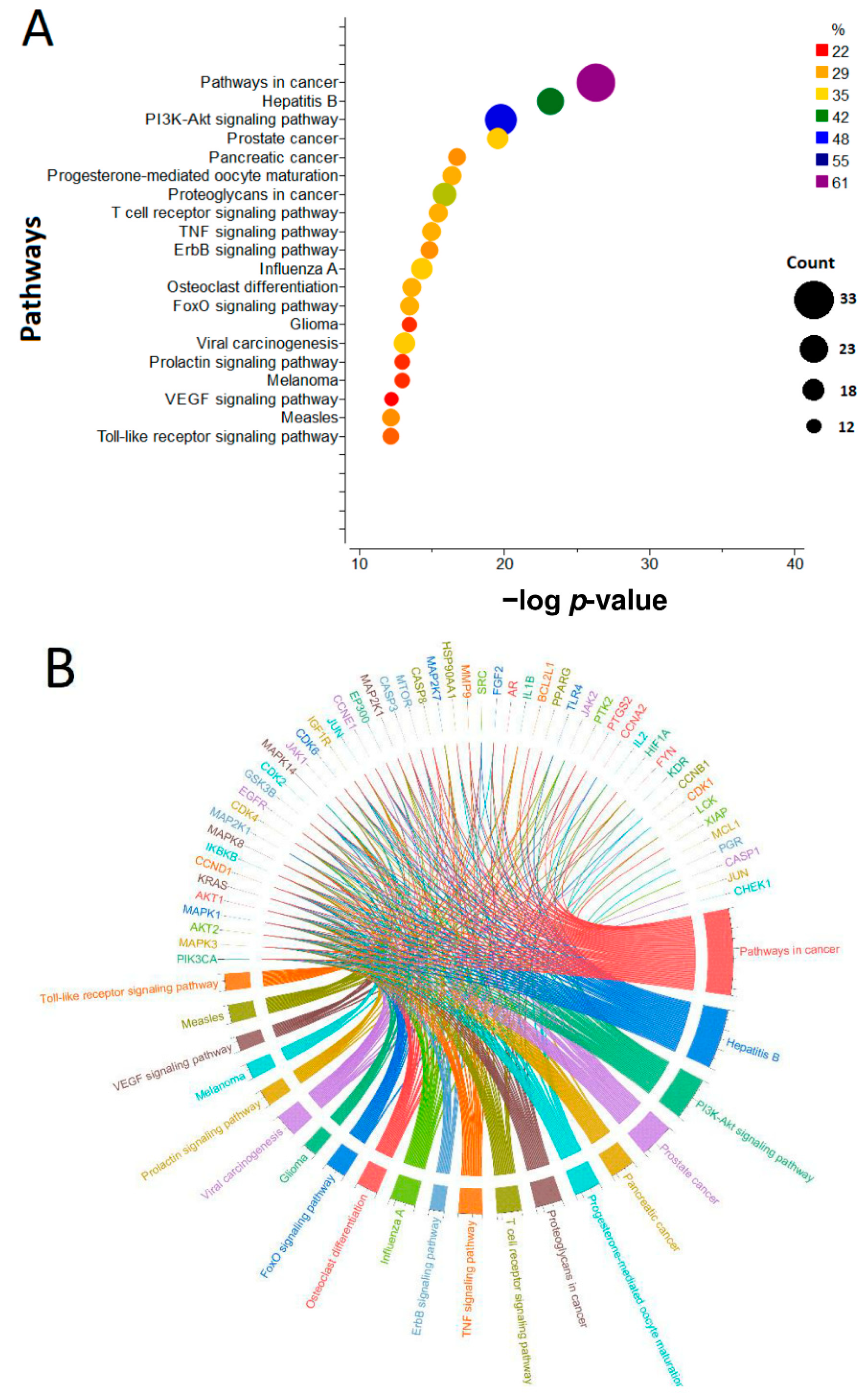
3.7.2. KEGG Pathway Enrichment Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schafer, K.A. The cell cycle: A review. Vet Pathol 1998, 35, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Coffman, J.A. Cell cycle development. Dev. Cell 2004, 6, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, A. Cell cycle checkpoints. Curr. Opin. Cell. Biol. 1994, 6, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Kar, S. Unraveling Cell-Cycle Dynamics in Cancer. Cell Syst. 2016, 2, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, E.S.; Singh, A.T.K. Cell-cycle Checkpoints and Aneuploidy on the Path to Cancer. In Vivo 2018, 32, 1–5. [Google Scholar]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Sofi, S.; Mehraj, U.; Qayoom, H.; Aisha, S.; Asdaq, S.M.B.; Almilaibary, A.; Mir, M.A. Cyclin-dependent kinases in breast cancer: Expression pattern and therapeutic implications. Med. Oncol. 2022, 39, 106. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.; Tsai, L.-H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [Green Version]
- Bury, M.; Calvé, B.L.; Ferbeyre, G.; Blank, V.; Lessard, F. New Insights into CDK Regulators: Novel Opportunities for Cancer Therapy. Trends Cell Biol. 2021, 31, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Pandey, S.; Djibo, R.; Darracq, A.; Calendo, G.; Zhang, H.; Henry, R.A.; Andrews, A.J.; Baylin, S.B.; Madzo, J.; Najmanovich, R.; et al. Selective CDK9 Inhibition by Natural Compound Toyocamycin in Cancer Cells. Cancers 2022, 14, 3340. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr. Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.C.; Morales, F.; Boffo, S.; Giordano, A. CDK9: A key player in cancer and other diseases. J. Cell Biochem. 2018, 119, 1273–1284. [Google Scholar] [CrossRef]
- Gordon, V.; Bhadel, S.; Wunderlich, W.; Zhang, J.; Ficarro, S.B.; Mollah, S.A.; Shabanowitz, J.; Hunt, D.F.; Xenarios, I.; Hahn, W.C.; et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol. Endocrinol. 2010, 24, 2267–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A.; Alfano, L. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef] [Green Version]
- Kretz, A.-L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumour Biol. 2017, 39, 1010428317694304. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019, 33, 5990–6000. [Google Scholar] [CrossRef]
- Krystof, V.; Baumli, S.; Fürst, R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr. Pharm. Des. 2012, 18, 2883–2890. [Google Scholar] [CrossRef] [Green Version]
- Mueller, D.; García-Cuéllar, M.-P.; Bach, C.; Buhl, S.; Maethner, E.; Slany, R.K. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7, e1000249. [Google Scholar] [CrossRef] [Green Version]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, C.N.; Mongroo, P.S.; Rich, A.S.; Schupp, M.; Bowser, M.J.; deLemos, A.S.; Tobias, J.W.; Liu, Y.; Hannigan, G.E.; Rustgi, A.K. Parvin-beta inhibits breast cancer tumorigenicity and promotes CDK9-mediated peroxisome proliferator-activated receptor gamma 1 phosphorylation. Mol. Cell Biol. 2008, 28, 687–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Gao, W.; Hu, F.; Xu, Z.; Wang, F. MicroRNA-874 inhibits cell proliferation and induces apoptosis in human breast cancer by targeting CDK9. FEBS Lett. 2014, 588, 4527–4535. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P.; Yang, R.-M.; Sutton, J.; Ramsay, R.G.; Gonda, T.J. CDK9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget 2016, 7, 9069–9083. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Investig. N. Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Walsby, E.; Pratt, G.; Shao, H.; Abbas, A.Y.; Fischer, P.M.; Bradshaw, T.D.; Brennan, P.; Fegan, C.; Wang, S.; Pepper, C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2014, 5, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Biessen, D.A.; Burger, H.; de Bruijin, P.; Lamers, C.H.J.; Naus, N.; Loferer, H.; Wiemer, E.A.C.; Mathijssen, R.H.J.; de Jonge, M.J.A. Phase I study of RGB-286638, a novel, multitargeted cyclin-dependent kinase inhibitor in patients with solid tumors. Clin. Cancer Res. 2014, 20, 4776–4783. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.-G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; Martin, M.S.; Pop-Damkov, P.; Su, N.; Franklin, V.N.R.; Chilamakuri, C.S.R.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef] [Green Version]
- McInnes, C. Progress in the evaluation of CDK inhibitors as anti-tumor agents. Drug Discov. Today 2008, 13, 875–881. [Google Scholar] [CrossRef]
- Zhai, S.; Senderowicz, A.M.; A Sausville, E.; Figg, W.D. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in clinical development. Ann. Pharmacother. 2002, 36, 905–911. [Google Scholar] [CrossRef]
- Lücking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. Chem. Med. Chem. 2017, 12, 1776–1793. [Google Scholar] [CrossRef]
- Di Meo, F.; Fabre, G.; Berka, K.; Ossman, T.; Chantemargue, B.; Paloncýová, M.; Marquet, P.; Otyepka, M.; Trouillas, P. In silico pharmacology: Drug membrane partitioning and crossing. Pharmacol. Res. 2016, 111, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.S.; Lopez-Cara, L.C.; Salvador, M.K. In silico pharmacology for a multidisciplinary drug discovery process. Drug Metab. Drug Interact. 2012, 27, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, C.; Priya, D.; Chen, L.; Zhu, H. Evaluating protein-protein interaction (PPI) networks for diseases pathway, target discovery, and drug-design using ‘in silico pharmacology’. Curr. Protein Pept. Sci. 2014, 15, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Mestres, J. Computational chemogenomics approaches to systematic knowledge-based drug discovery. Curr. Opin. Drug Discov. Dev. 2004, 7, 304–313. [Google Scholar]
- Pérot, S.; Regad, L.; Reynès, C.; Spérandio, O.; Miteva, M.A.; Villoutreix, B.O.; Camproux, A.-C. Insights into an Original Pocket-Ligand Pair Classification: A Promising Tool for Ligand Profile Prediction. PLoS ONE 2013, 8, e63730. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.N.H.A.C.J.W.C.H.; Huang, C.; Wang, C. The Anti-Tumor Effect and Mechanisms of Action of Penta-Acetyl Geniposide. Curr. Cancer Drug Targets 2005, 5, 299–305. [Google Scholar] [CrossRef]
- Cai, L.; Li, C.-M.; Tang, W.-J.; Liu, M.-M.; Chen, W.-N.; Qiu, Y.-Y.; Li, R. Therapeutic Effect of Penta-acetyl Geniposide on Adjuvant-Induced Arthritis in Rats: Involvement of Inducing Synovial Apoptosis and Inhibiting NF-κB Signal Pathway. Inflammation 2018, 41, 2184–2195. [Google Scholar] [CrossRef]
- Peng, C.-H.; Tseng, T.-H.; Huang, C.-N.; Hsu, S.-P.; Wang, C.-J. Apoptosis induced by penta-acetyl geniposide in C6 glioma cells is associated with JNK activation and Fas ligand induction. Toxicol. Appl. Pharmacol. 2005, 202, 172–179. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Hsu, H.-Y.; Yang, J.-J.; Lin, S.-Y.; Lin, C.-C. Comparisons of geniposidic acid and geniposide on antitumor and radioprotection after sublethal irradiation. Cancer Lett. 1997, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Sagara, K.; Yoshida, T.; Tong, Y.Y.; Zhang, G.D.; Chen, Y.H. Determination of geniposide, gardenoside, geniposidic acid and genipin-1-beta-gentiobioside in Gardenia jasminoides by high-performance liquid chromatography. J. Chromatogr. 1988, 455, 410–414. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Tseng, T.-H.; Lee, M.-J.; Hsu, J.-D.; Wang, C.-J. Induction of apoptosis by penta-acetyl geniposide in rat C6 glioma cells. Chem. Interact. 2002, 141, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.-H.; Huang, C.-N.; Hsu, S.-P.; Wang, C.-J. Penta-acetyl geniposide-induced apoptosis involving transcription of NGF/p75 via MAPK-mediated AP-1 activation in C6 glioma cells. Toxicology 2007, 238, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Tseng, T.H.; Lin, J.K. Penta-acetyl geniposide: Isolation, identification and primary effect on C6 glioma cells in vitro. Anticancer Res. 1992, 12, 911–915. [Google Scholar]
- Peng, C.H.; Peng, C.-H.; Huang, C.-N.; Hsu, S.-P.; Wang, C.-J. Penta-acetyl geniposide induce apoptosis in C6 glioma cells by modulating the activation of neutral sphingomyelinase-induced p75 nerve growth factor receptor and protein kinase Cdelta pathway. Mol. Pharmacol. 2006, 70, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Lentini, G. Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin. Biomedicines 2018, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef]
- Davoodvandi, A.; Varkani, M.S.; Clark, C.C.T.; Jafarnejad, S. Quercetin as an anticancer agent: Focus on esophageal cancer. J. Food Biochem. 2020, 44, e13374. [Google Scholar] [CrossRef]
- Niedzwiecki, A.; Roomi, M.W.; Kalinovsky, T.; Rath, M. Anticancer Efficacy of Polyphenols and Their Combinations. Nutrients 2016, 8, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soofiyani, S.R.; Hosseini, K.; Forouhandeh, H.; Ghasemnejad, T.; Tarhriz, V.; Asgharian, P.; Reiner, Ž.; Sharifi-Rad, J.; Cho, W.C. Quercetin as a Novel Therapeutic Approach for Lymphoma. Oxid. Med. Cell Longev. 2021, 2021, 3157867. [Google Scholar] [CrossRef] [PubMed]
- Hashemzaei, M.; Delarami Far, A.; Yari, A.; Heravi, R.E.; Tabrizian, K.; Taghdisi, S.M.; Sadegh, S.E.; Tsarouhas, K.; Kouretas, D.; Tzanakakis, G.; et al. Anticancer and apoptosis-inducing effects of quercetin in vitro and in vivo. Oncol. Rep. 2017, 38, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinayak, M.; Maurya, A.K. Quercetin Loaded Nanoparticles in Targeting Cancer: Recent Development. Anti Cancer Agents Med. Chem. 2019, 19, 1560–1576. [Google Scholar] [CrossRef]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef]
- Hosseini, A.; Razavi, B.M.; Banach, M.; Hosseinzadeh, H. Quercetin and metabolic syndrome: A review. Phytotherapy Res. 2021, 35, 5352–5364. [Google Scholar] [CrossRef]
- Czerwonka, A.; Maciolek, U.; Kalafut, J.; Mendyk, E.; Kuzniar, A.; Rzeski, W. Anticancer effects of sodium and potassium quercetin-5′-sulfonates through inhibition of proliferation, induction of apoptosis, and cell cycle arrest in the HT-29 human adenocarcinoma cell line. Bioorg. Chem. 2020, 94, 103426. [Google Scholar] [CrossRef]
- Shafabakhsh, R.; Asemi, Z. Quercetin: A natural compound for ovarian cancer treatment. J. Ovar. Res. 2019, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Massi, A.; Bortolini, O.; Ragno, D.; Bernardi, T.; Sacchetti, G.; Tacchini, M.; De Risi, C. Research Progress in the Modification of Quercetin Leading to Anticancer Agents. Molecules 2017, 22, 1270. [Google Scholar] [CrossRef]
- Tang, S.-M.; Deng, X.-T.; Zhou, J.; Li, Q.-P.; Ge, X.-X.; Miao, L. Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed. Pharmacother. 2019, 121, 109604. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, J.; Li, X.; Wu, Y.; Shi, H.; Chen, Y.; Lu, G.; Shen, H.; Lu, G.; Zhou, J. Quercetin induces p53-independent cancer cell death through lysosome activation by the transcription factor EB and Reactive Oxygen Species-dependent ferroptosis. Br. J. Pharmacol. 2020, 178, 1133–1148. [Google Scholar] [CrossRef]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Joshi, S.; Semwal, D.; Bisht, A.; Paliwal, S.; Dwivedi, J.; Sharma, S. Curcumin: An Insight into Molecular Pathways Involved in Anticancer Activity. Mini-Reviews Med. Chem. 2021, 21, 2420–2457. [Google Scholar] [CrossRef] [PubMed]
- Unlu, A.; Nayir, E.; Kalenderoglu, M.D.; Kirca, O.; Ozdogan, M. Curcumin (Turmeric) and cancer. J. BUON 2016, 21, 1050–1060. [Google Scholar] [PubMed]
- Zia, A.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Samarghandian, S. The role of curcumin in aging and senescence: Molecular mechanisms. Biomed. Pharmacother. 2021, 134, 111119. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.C.; Kumar, N.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer Activity of Curcumin and Its Analogues: Preclinical and Clinical Studies. Cancer Investig. 2017, 35, 1–22. [Google Scholar] [CrossRef]
- Hassanalilou, T.; Ghavamzadeh, S.; Khalili, L. Curcumin and Gastric Cancer: A Review on Mechanisms of Action. J. Gastrointest. Cancer 2019, 50, 185–192. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [Green Version]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, Pharmaceutical, Nutraceutical, and Analytical Aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Kamarudin, M.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, J.; Cui, R.; Lin, J.; Ding, X. Curcumin in Treating Breast Cancer: A Review. J. Lab. Autom. 2016, 21, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Mbese, Z.; Khwaza, V.; Aderibigbe, B.A. Curcumin and Its Derivatives as Potential Therapeutic Agents in Prostate, Colon and Breast Cancers. Molecules 2019, 24, 4386. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.-J.; Tang, J.-Y.; Lin, L.-C.; Lien, W.-J.; Cheng, Y.-B.; Chang, F.-R.; Ou-Yang, F.; Chang, H.-W. Withanolide C Inhibits Proliferation of Breast Cancer Cells via Oxidative Stress-Mediated Apoptosis and DNA Damage. Antioxidants 2020, 9, 873. [Google Scholar] [CrossRef]
- Xu, Q.Q.; Wang, K.W. Natural Bioactive New Withanolides. Mini Rev. Med. Chem. 2020, 20, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; He, J.-X.; Hu, H.-X.; Zhang, K.; Wang, X.-N.; Zhao, B.-B.; Lou, H.-X.; Ren, D.-M.; Shen, T. Withanolides from the genus Physalis: A review on their phytochemical and pharmacological aspects. J. Pharm. Pharmacol. 2020, 72, 649–669. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Cheng, Y.-B.; Chuang, Y.-T.; Yang, K.-H.; Chang, F.-R.; Liu, W.; Chang, H.-W. Oxidative Stress and AKT-Associated Angiogenesis in a Zebrafish Model and Its Potential Application for Withanolides. Cells 2022, 11, 961. [Google Scholar] [CrossRef]
- Xia, G.Y.; Cao, S.-J.; Chen, L.-X.; Qiu, F. Natural withanolides, an update. Nat. Prod. Rep. 2022, 39, 784–813. [Google Scholar] [CrossRef]
- Xiang, K.; Li, C.; Li, M.-X.; Song, Z.-R.; Ma, X.-X.; Sun, D.-J.; Li, H.; Chen, L.-X. Withanolides isolated from Tubocapsicum anomalum and their antiproliferative activity. Bioorg. Chem. 2021, 110, 104809. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Huang, S.; Chen, Z.; Wang, H.; Wu, H.; Zhang, D. Temsirolimus, the mTOR inhibitor, induces autophagy in adenoid cystic carcinoma: In vitro and in vivo. Pathol. Res. Pract. 2014, 210, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Perola, E.; Walters, W.P.; Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins Struct. Funct. Bioinform. 2004, 56, 235–249. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A. Prediction of ADMET Properties. Chem. Med. Chem. 2006, 1, 920–937. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Khafaji, K.A.; Tok, T.T. Amygdalin as multi-target anticancer drug against targets of cell division cycle: Double docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020, 39, 1965–1974. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, U.; Blairy, S.; Kleck, R.E. The Intensity of Emotional Facial Expressions and Decoding Accuracy. J. Nonverbal Behav. 1997, 21, 241–257. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef]
- Khan, R.A.; Hossain, R.; Siyadatpanah, A.; Al-Khafaji, K.; Khalipha, A.B.R.; Dey, D.; Asha, U.H.; Biswas, P.; Saikat, A.S.M.; Chenari, H.A.; et al. Diterpenes/Diterpenoids and Their Derivatives as Potential Bioactive Leads against Dengue Virus: A Computational and Network Pharmacology Study. Molecules 2021, 26, 6821. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
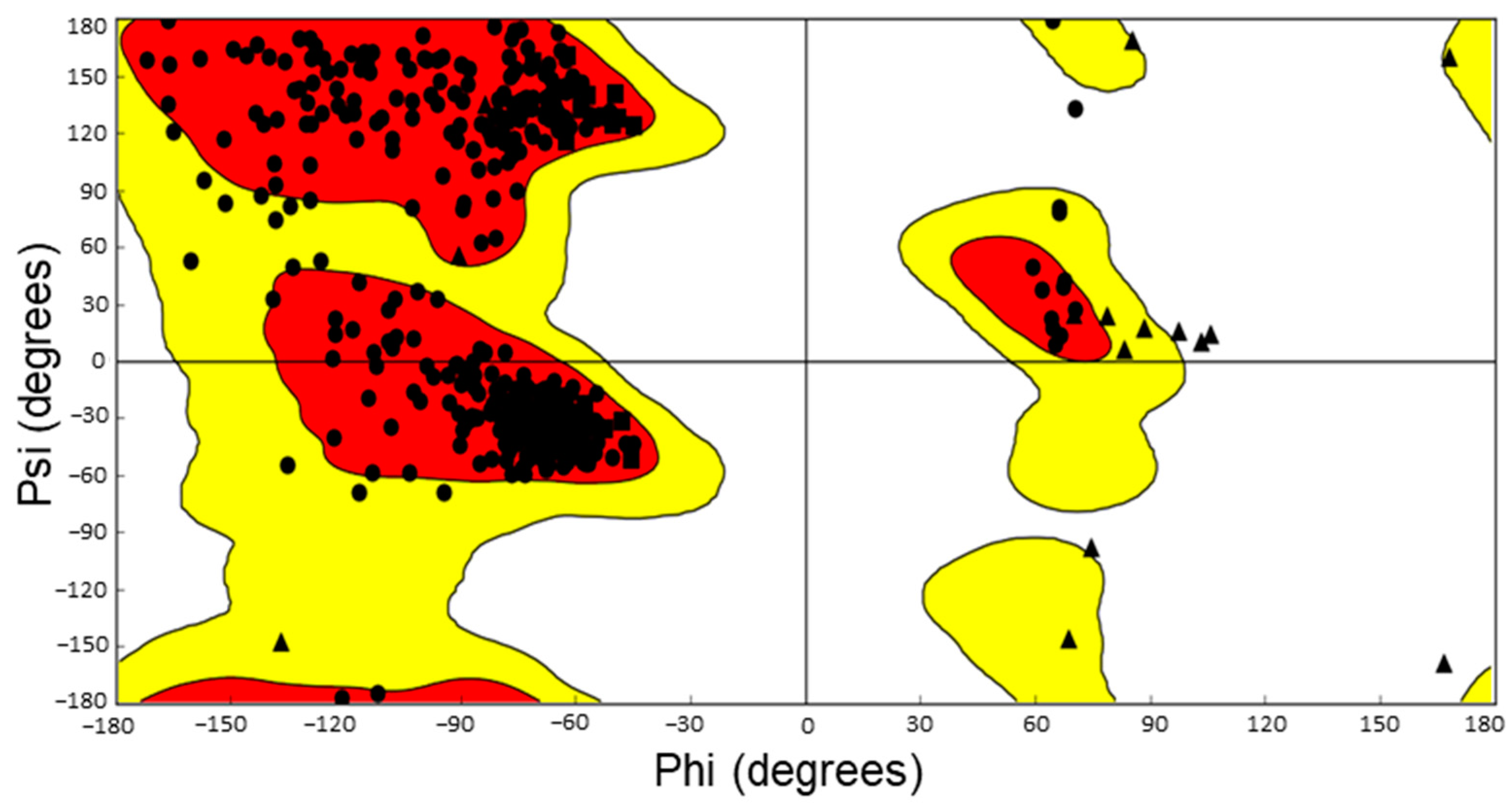
- Sheik, S.; Sundararajan, P.; Hussain, A.; Sekar, K. Ramachandran plot on the web. Bioinformatics 2002, 18, 1548–1549. [Google Scholar] [CrossRef] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2022, 8, 5. [Google Scholar]
- Saikat, A.S.M.; Uddin, E.; Ahmad, T.; Mahmud, S.; Imran, A.S.; Ahmed, S.; Alyami, S.A.; Moni, M.A. Structural and Functional Elucidation of IF-3 Protein of Chloroflexus aurantiacus Involved in Protein Biosynthesis: An In Silico Approach. BioMed Res. Int. 2021, 2021, 9050026. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K.; Marwaha, T.K. Molecular Docking in Formulation and Development. Curr. Cancer Drug Targets 2019, 16, 30–39. [Google Scholar] [CrossRef]
- Saikat, A.S.M.; Islam, R.; Mahmud, S.; Imran, M.A.S.; Alam, M.S.; Masud, M.H.; Uddin, M.E. Structural and Functional Annotation of Uncharacterized Protein NCGM946K2_146 of Mycobacterium Tuberculosis: An In-Silico Approach. Proceedings 2020, 66, 13. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [Green Version]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Ferreira, L.L.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. Chem. Med. Chem. 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine Towards Cancer: 111In-Labeled Nanoparticles. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Bruno, C.D.; Harmatz, J.S.; Duan, S.X.; Zhang, Q.; Chow, C.R.; Greenblatt, D.J. Effect of lipophilicity on drug distribution and elimination: Influence of obesity. Br. J. Clin. Pharmacol. 2021, 87, 3197–3205. [Google Scholar] [CrossRef] [PubMed]
- Yilancioglu, K.; Weinstein, Z.B.; Meydan, C.; Akhmetov, A.; Toprak, I.; Durmaz, A.; Iossifov, I.; Kazan, H.; Roth, F.P.; Cokol, M. Target-Independent Prediction of Drug Synergies Using Only Drug Lipophilicity. J. Chem. Inf. Model. 2014, 54, 2286–2293. [Google Scholar] [CrossRef]
- Paneth, A.; Hawrył, A.; Plech, T.; Hawrył, M.; Świeboda, R.; Janowska, D.; Wujec, M.; Paneth, P. Lipophilicity Studies on Thiosemicarbazide Derivatives. Molecules 2017, 22, 952. [Google Scholar] [CrossRef] [Green Version]
- Van der Spoel, D.; Manzetti, S.; Zhang, H.; Klamt, A. Prediction of Partition Coefficients of Environmental Toxins Using Computational Chemistry Methods. ACS Omega 2019, 4, 13772–13781. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, T.J.; Macdonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. Increasing small molecule drug developability in sub-optimal chemical space. Med. Chem. Comm. 2013, 4, 673–680. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Turfus, S.; Delgoda, R.; Picking, D.; Gurley, B.J. Chapter 25—Pharmacokinetics; Academic Press: Boston, MA, USA, 2017; pp. 495–512. [Google Scholar]
- Saghir, S.A.; Ansari, R.A. Pharmacokinetics, in Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Waller, D.G.; Sampson, A.P. 2-Pharmacokinetics. In Medical Pharmacology and Therapeutics, 5th ed.; Waller, D.G., Sampson, A.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 33–62. [Google Scholar]
- Paixão, P.; Gouveia, L.F.; Morais, J.A. Prediction of the human oral bioavailability by using in vitro and in silico drug related parameters in a physiologically based absorption model. Int. J. Pharm. 2012, 429, 84–98. [Google Scholar] [CrossRef]
- Wu, X.-X.; Huang, X.-L.; Chen, R.-R.; Li, T.; Ye, H.-J.; Xie, W.; Huang, Z.-M.; Cao, G.-Z. Paeoniflorin Prevents Intestinal Barrier Disruption and Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in Caco-2 Cell Monolayers. Inflammation 2019, 42, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Henri, J.; Lanceleur, R.; Delmas, J.-M.; Fessard, V.; Huguet, A. Permeability of the Cyanotoxin Microcystin-RR across a Caco-2 Cells Monolayer. Toxins 2021, 13, 178. [Google Scholar] [CrossRef]
- Vincze, A.; Dargó, G.; Rácz, A.; Balogh, G.T. A corneal-PAMPA-based in silico model for predicting corneal permeability. J. Pharm. Biomed. Anal. 2021, 203, 114218. [Google Scholar] [CrossRef] [PubMed]
- Angelis, I.D.; Turco, L. Caco-2 cells as a model for intestinal absorption. Curr. Protoc. Toxicol. 2011, 47, 20.6.1–20.6.15. [Google Scholar] [CrossRef]
- Volpe, D.A. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med. Chem. 2011, 3, 2063–2077. [Google Scholar] [CrossRef]
- Drago, S.; El Asmar, R.; Di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J. Gastroenterol. 2006, 41, 408–419. [Google Scholar] [CrossRef]
- Volpe, D.A. Advances in cell-based permeability assays to screen drugs for intestinal absorption. Expert Opin. Drug Discov. 2020, 15, 539–549. [Google Scholar] [CrossRef]
- Le Roux, A.; Blaise, E.; Boudreault, P.-L.; Comeau, C.; Doucet, A.; Giarrusso, M.; Collin, M.-P.; Neubauer, T.; Koelling, F.; Göller, A.H.; et al. Structure–Permeability Relationship of Semipeptidic Macrocycles—Understanding and Optimizing Passive Permeability and Efflux Ratio. J. Med. Chem. 2020, 63, 6774–6783. [Google Scholar] [CrossRef]
- Toutain, P.L.; Bousquet-Mélou, A. Plasma clearance. J. Vet Pharmacol. Ther. 2004, 27, 415–425. [Google Scholar] [CrossRef]
- Roth, R.A.; Wiersma, D.A. Role of the Lung in Total Body Clearance of Circulating Drugs. Clin. Pharmacokinet. 1979, 4, 355–367. [Google Scholar] [CrossRef]
- Pippa, L.F.; de Oliveira, M.L.; Rocha, A.; de Andrade, J.M.; Lanchote, V.L. Total, renal and hepatic clearances of doxorubicin and formation clearance of doxorubicinol in patients with breast cancer: Estimation of doxorubicin hepatic extraction ratio. J. Pharm. Biomed. Anal. 2020, 185, 113231. [Google Scholar] [CrossRef]
- Mathialagan, S.; Piotrowski, M.A.; Tess, D.A.; Feng, B.; Litchfield, J.; Varma, M.V. Quantitative Prediction of Human Renal Clearance and Drug-Drug Interactions of Organic Anion Transporter Substrates Using In Vitro Transport Data: A Relative Activity Factor Approach. Drug Metab. Dispos. 2017, 45, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.; de Bruyn, T.; Darwich, A.S.; Houston, J.B. Simultaneous Assessment In Vitro of Transporter and Metabolic Processes in Hepatic Drug Clearance: Use of a Media Loss Approach. Drug Metab. Dispos. 2018, 46, 405–414. [Google Scholar] [CrossRef]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Soulières, D.; Gelmon, K.A. Sotorasib: Is Maximum Tolerated Dose Really the Issue at Hand? J. Clin. Oncol. 2021, 39, 3427–3429. [Google Scholar] [CrossRef]
- Stampfer, H.G.; Gabb, G.M.; Dimmitt, S.B. Why maximum tolerated dose? Br. J. Clin. Pharmacol. 2019, 85, 2213–2217. [Google Scholar] [CrossRef]
- Bozkurt, B. Target Dose Versus Maximum Tolerated Dose in Heart Failure: Time to Calibrate and Define Actionable Goals. JACC Heart Fail 2019, 7, 359–362. [Google Scholar] [CrossRef]
- Kojima, M. Adaptive design for identifying maximum tolerated dose early to accelerate dose-finding trial. BMC Med. Res. Methodol. 2022, 22, 97. [Google Scholar] [CrossRef]
- Gad, S.C. LD50/LC50 (Lethal Dosage 50/Lethal Concentration 50). In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 58–60. [Google Scholar]
- Aucar, M.G.; Cavasotto, C.N. Molecular Docking Using Quantum Mechanical-Based Methods. Methods Mol. Biol. 2020, 2114, 269–284. [Google Scholar]
- Cavasotto, C.; Aucar, M.G. High-Throughput Docking Using Quantum Mechanical Scoring. Front. Chem. 2020, 8, 246. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Kassel, D.B. Applications of high-throughput ADME in drug discovery. Curr. Opin. Chem. Biol. 2004, 8, 339–345. [Google Scholar] [CrossRef]
- Wang, J.; Skolnik, S. Recent Advances in Physicochemical and ADMET Profiling in Drug Discovery. Chem. Biodivers. 2009, 6, 1887–1899. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Liu, G.; Tang, Y. In Silico ADMET Prediction: Recent Advances, Current Challenges and Future Trends. Curr. Top. Med. Chem. 2013, 13, 1273–1289. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H. Improving compound quality through in vitro and in silico physicochemical profiling. Chem. Biodivers. 2009, 6, 1760–1766. [Google Scholar] [CrossRef]
- Cruciani, G.; Milletti, F.; Storchi, L.; Sforna, G.; Goracci, L. ChemInform Abstract: In silico pKa Prediction and ADME Profiling. ChemInform 2010, 6, 1812–1821. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Prasanna, S.; Doerksen, R.J. Topological polar surface area: A useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Fernandes, J.; Gattass, C.R. Topological polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J. Med. Chem. 2009, 52, 1214–1218. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2019, 25, 248–258. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Augustijns, P.; Wuyts, B.; Hens, B.; Annaert, P.; Butler, J.; Brouwers, J. A review of drug solubility in human intestinal fluids: Implications for the prediction of oral absorption. Eur. J. Pharm. Sci. 2014, 57, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, A.Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Zhang, Y. Overview of Transporters in Pharmacokinetics and Drug Discovery. Curr. Protoc. Pharmacol. 2018, 82, e46. [Google Scholar] [CrossRef]
- Reichel, A.; Lienau, P. Pharmacokinetics in Drug Discovery: An Exposure-Centred Approach to Optimising and Predicting Drug Efficacy and Safety. Handb. Exp. Pharmacol. 2016, 232, 235–260. [Google Scholar]
- Glassman, P.M.; Balthasar, J.P. Physiologically-based modeling of monoclonal antibody pharmacokinetics in drug discovery and development. Drug Metab. Pharmacokinet. 2019, 34, 3–13. [Google Scholar] [CrossRef]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction--Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef] [PubMed]
- Yew, W.W.; Chang, K.C.; Chan, D.P. Oxidative Stress and First-Line Antituberculosis Drug-Induced Hepatotoxicity. Antimicrob. Agents Chemother. 2018, 62, e02637-17. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Schiano, T.D. Review article: Drug hepatotoxicity. Aliment. Pharmacol. Ther. 2007, 25, 1135–1151. [Google Scholar] [CrossRef]
- Murray, K.F.; Hadzic, N.; Wirth, S.; Bassett, M.; Kelly, D. Drug-related Hepatotoxicity and Acute Liver Failure. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 395–405. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Saikia, S.; Bordoloi, M. Molecular Docking: Challenges, Advances and its Use in Drug Discovery Perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Guilbert, C.; James, T.L. Docking to RNA via Root-Mean-Square-Deviation-Driven Energy Minimization with Flexible Ligands and Flexible Targets. J. Chem. Inf. Model. 2008, 48, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Rout, A.K.; Barnwal, R.P.; Agarwal, G.; Chary, K.V.R. Root-mean-square-deviation-based rapid backbone resonance assignments in proteins. Org. Magn. Reson. 2010, 48, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Shigeta, Y. Temperature-Shuffled Structural Dissimilarity Sampling Based on a Root-Mean-Square Deviation. J. Chem. Inf. Model. 2018, 58, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Cazals, F.; Tetley, R. Characterizing molecular flexibility by combining least root mean square deviation measures. Proteins: Struct. Funct. Bioinform. 2019, 87, 380–389. [Google Scholar] [CrossRef]
- Pitera, J.W. Expected Distributions of Root-Mean-Square Positional Deviations in Proteins. J. Phys. Chem. B 2014, 118, 6526–6530. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, T.; Karplus, M. Thermodynamics of protein folding: A microscopic view. Biophys. Chem. 2003, 100, 367–395. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.-H.; Ham, S. Distinct Role of Hydration Water in Protein Misfolding and Aggregation Revealed by Fluctuating Thermodynamics Analysis. Accounts Chem. Res. 2015, 48, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Ham, S. Examining a Thermodynamic Order Parameter of Protein Folding. Sci. Rep. 2018, 8, 7148. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Chen, H.; Wang, X.; Chai, H.; Shao, X.; Cai, W.; Chipot, C. Finding an Optimal Pathway on a Multidimensional Free-Energy Landscape. J. Chem. Inf. Model. 2020, 60, 5366–5374. [Google Scholar] [CrossRef]
- Fu, H.; Shao, X.; Cai, W.; Chipot, C. Taming Rugged Free Energy Landscapes Using an Average Force. Accounts Chem. Res. 2019, 52, 3254–3264. [Google Scholar] [CrossRef]
- Chandran, U.; Mehendale, N.; Tillu, G.; Patwardhan, B. Network Pharmacology of Ayurveda Formulation Triphala with Special Reference to Anti-Cancer Property. Comb. Chem. High Throughput Screen. 2015, 18, 846–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakle, N.S.; More, S.A.; Mokale, S.N. A network pharmacology-based approach to explore potential targets of Caesalpinia pulcherima: An updated prototype in drug discovery. Sci. Rep. 2020, 10, 17217. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C. Targeting protein-protein interactions for drug discovery. Methods Mol. Biol. 2015, 1278, 93–106. [Google Scholar] [PubMed]
- Fry, D.C. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84, 535–552. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Y.-H.; Lu, G.; Huang, T.; Cai, Y.-D. Analysis of cancer-related lncRNAs using gene ontology and KEGG pathways. Artif. Intell. Med. 2017, 76, 27–36. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein–Ligand Complex | Docking Score (kcal/mol) | H-Bonds | Non-Bonding Interactions |
|---|---|---|---|
| 6GZH-LCI complex (Co-crystalized ligand) | −11.425 | CYS106, ASP109 | Polar: GLN27, HIS108 Hydrophobic: ILE25, PHE30, ALA166, VAL79, ALA46, PHE103, PHE105, CYS106, LEU156 Charged (Negative): ASP167, ASP104, GLU107, ASP109 Charged (Positive): LYS48 Glycine: GLY26 |
| 6GZH-Geniposidic Acid complex | −13.908 | ILE25, ALA153, ASN154, ASP104, CYS106 | Hydrophobic: ALA153, LEU156, PHE30, VAL33, PHE168, ALA166, ALA46, VAL79, PHE103, PHE105 Charged (Negative): ASP167, ASP109 Charged (Positive): LYS151, LYS48 Glycine: GLY26 |
| 6GZH-Quercetin complex | −10.775 | CYS106, GLU107 | Polar: HIS108 Hydrophobic: LEU156, PHE30, VAL33, VAL79, ALA46, PHE103, PHE105, ILE25 Charged (Negative): ASP167, GLU107, ASP109 Charged (Positive): LYS48 |
| 6GZH-Geniposide complex | −9.969 | ASN154, CYS106, ASP109 | Polar: HIS108 Hydrophobic: LEU156, ALA25, ALA166, PHE30, VAL33, ALA46, VAL79, PHE103, PHE105 Charged (Negative): ASP167, ASP104, GLU107 Charged (Positive): LYS48 |
| 6GZH-Curcumin complex | −9.898 | LYS48, CYS106, ASN154 | Hydrophobic: ALA166, PHE168, VAL33, PHE30, ALA46, PHE103, PHE105, CYS106, LEU156, ILE25, VAL79, LEU70 Charged (Negative): ASP109, ASP167, GLU66 Charged (Positive): LYS151 Glycine: GLY26 |
| 6GZH-Withanolide C complex | −8.114 | ILE25, GLN27, ALA153, LYS48, ASP109 | Polar: ASN154 Hydrophobic: PHE30, VAL33, LEU156, ALA46, ALA166, PHE103, CYS106, VAL79 Charged (Negative): ASP167 Glycine: GLY26 |
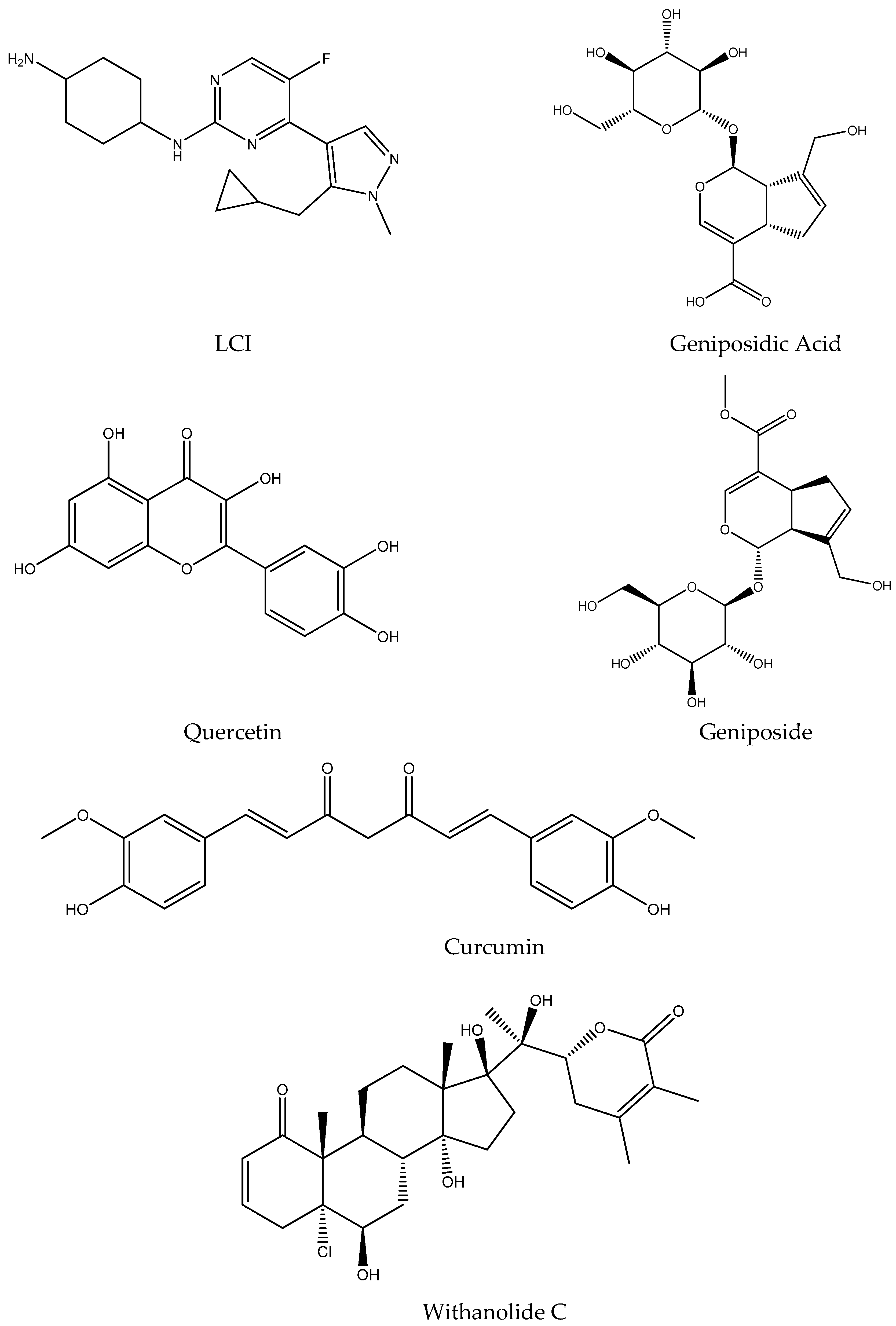
| LCI | Geniposidic Acid | Quercetin | Geniposide | Curcumin | Withanolide C | |
|---|---|---|---|---|---|---|
| Formula | C18H25FN6 | C16H22O10 | C15H10O7 | C17H24O10 | C21H20O6 | C28H39ClO7 |
| Molecular weight (g/mol) | 344.43 | 374.34 | 302.24 | 388.37 | 368.38 | 523.06 |
| H-Bond acceptors | 5 | 10 | 7 | 10 | 6 | 7 |
| H-Bond donors | 2 | 6 | 5 | 5 | 2 | 4 |
| Num. rotatable bonds | 5 | 5 | 1 | 6 | 8 | 2 |
| TPSA (Å2) | 81.65 | 166.14 | 131.36 | 155.14 | 93.06 | 124.29 |
| Fraction Csp3 | 0.61 | 0.69 | 0.00 | 0.71 | 0.14 | 0.79 |
| Molar refractivity | 95.68 | 82.57 | 78.03 | 86.89 | 102.80 | 135.75 |
| LogPo/w (XLOGP3) | 2.40 | −2.67 | 1.54 | −2.34 | 3.20 | 2.03 |
| LogS (ESOL) | −3.48 | −0.15 | −3.16 | −0.38 | −3.94 | −4.23 |
| Max. tolerated dose (human) (log mg/kg/day) | −0.142 | 1.339 | 0.499 | 0.528 | 0.081 | −0.694 |
| Oral rat acute toxicity (LD50 mol/kg) | 2.37 | 2.085 | 2.471 | 2.188 | 1.833 | 2.417 |
| Hepatotoxicity | Yes | No | No | No | No | No |
| Minnow toxicity (log mM) | 1.21 | 6.62 | 3.721 | 6.612 | −0.081 | 2.52 |
| Blood–brain barrier (log BB) | −0.162 | −1.041 | −1.098 | −1.281 | −0.562 | −1.004 |
| Caco-2 permeability | 1.344 | −0.505 | −0.229 | 0.35 | −0.093 | 0.682 |
| Total clearance (log ml/min/kg) | 0.954 | 1.325 | 0.407 | 1.404 | −0.002 | 0.023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saikat, A.S.M.; Al-Khafaji, K.; Akter, H.; Choi, J.-G.; Hasan, M.; Lee, S.-S. Nature-Derived Compounds as Potential Bioactive Leads against CDK9-Induced Cancer: Computational and Network Pharmacology Approaches. Processes 2022, 10, 2512. https://doi.org/10.3390/pr10122512
Saikat ASM, Al-Khafaji K, Akter H, Choi J-G, Hasan M, Lee S-S. Nature-Derived Compounds as Potential Bioactive Leads against CDK9-Induced Cancer: Computational and Network Pharmacology Approaches. Processes. 2022; 10(12):2512. https://doi.org/10.3390/pr10122512
Chicago/Turabian StyleSaikat, Abu Saim Mohammad, Khattab Al-Khafaji, Hafeza Akter, Jong-Gu Choi, Mahbub Hasan, and Sang-Suk Lee. 2022. "Nature-Derived Compounds as Potential Bioactive Leads against CDK9-Induced Cancer: Computational and Network Pharmacology Approaches" Processes 10, no. 12: 2512. https://doi.org/10.3390/pr10122512