Fragrances are applied extensively as an attractive attribute in the formulation of many consumer products. The global flavours and fragrances (F&F) market size is expected to expand from the original value of USD 26.54 billion (2022) to USD 36.49 billion (2029) at a compounded annual growth rate of 4.7% [
1]. The demand for novel fragrance molecules in the industry is greater than ever due to the stricter safety and environmental (e.g., biodegradability) regulations, which have led to the obsolescence of some existing products [
2]. Unlike other senses, olfaction is poorly understood [
2]. The design of fragrance molecules still heavily depends on empirical methods, either referring to the knowledge from experts or through experiments. This trial-and-error approach is too tedious to allow the exploration of all potential candidates, as fragrance molecules have complex structures. Thus, there is a risk of missing better fragrance molecules that have the potential to be incorporated into consumer products [
3]. The conventional method is a resource-intensive process, which makes launching a new fragrance molecule costly and time-consuming [
2]. Moreover, most of the fragrances’ odour descriptions in established databases are reported without the indication of concentration [
2]. This could be another hurdle as the concentration of fragrance required in various products might be different.
To address the challenges involved in the design of fragrance molecules, a systematic framework should be developed for designing and screening suitable fragrances that fulfil the product’s requirement before experimental verification. Computer-aided molecular design (CAMD) approach is a potential tool for the screening and/or design of fragrance molecules by predicting the molecular structure using a set of desired sensorial and technical properties. However, a pre-requisite for the initiation of CAMD modelling is property predictive models. Perceived odours are determined by the structure of a fragrance molecule, the latter of which can be described using structural, geometrical, topological, physicochemical, and electronic descriptors [
4]. Hence, machine learning (ML) tools have the potential to develop prediction models by linking the molecular structure to properties using topological indices as the numerical representation of the structure.
1.1. Computer-Aided Molecular Design (CAMD)
CAMD is a reverse engineering approach to screening novel chemicals by combining structural groups systematically to yield high-performance molecules [
5]. In CAMD, property prediction models, such as group contribution (GC) models, are required. GC methods assume that the properties of a molecule can be estimated by the number of occurrences of different sub-structures, known as “groups”. In addition to GC methods, topological indices (TIs), one of the structural descriptors, were employed by the quantitative structure-property relationship (QSPR) for property estimation. Some of the common TIs, which include connectivity index, shape index, etc., can be used to differentiate very similar structures like isomers [
6].
CAMD is applied widely in various applications related to solvent design [
7] and integrated process and product design problems [
8]. In recent years, there have been several developments in the application of these tools in the field of product development as well. Liu et al. [
9] coupled ML-based atom contribution (MLAC) with CAMD to forecast the surface-charged density profile and construct a solvent for ibuprofen with improved economic, safety, health, and environmental aspects. An artificial neural network model was utilized to generate the structure-odour relationship (SOR) model for aromatic component mixtures by utilising the profiles of molecular surface charge density (r-profiles) as the descriptors [
10]. It was also employed for the identification of potential solvent candidates that allow bio-oil to satisfy targeted properties with minimal solvent addition [
11]. Moreover, Yee et al. [
12] developed a framework for personal care product design by incorporating safety, health, and performance aspects in CAMD. By imposing constraints for safety and health hazards in CAMD, molecules generated were less harmful while possessing excellent product performance. There are some recent works in the CAMD field related to fragrance products. MILP/MINLP models for the design and screening of fragrance in shampoo were developed by Zhang et al. [
3]. The CAMD model was utilised to remove the molecules that are out of the range of the constraints and properties of fragrant molecule design. In addition, fragrances in body lotion were modelled using rules generated with an enhanced hyperbox ML coupled with CAMD [
13]. The hybrid CAMD framework was able to produce a variety of viable compounds that met all structural and physical property requirements. In both works, CAMD was proven to be effective in developing potential fragrant molecules for consumer products. Comprehensive reviews of the latest developments in this field can be found in the review articles by Chemmangattuvalappil [
14] and Zhang et al. [
15].
A recent contribution has demonstrated that rough set-based machine learning (RSML) can be used to develop a model to predict the fragrance of molecules and used the developed model for identifying novel fragrant molecules [
16]. In this previous work, a single molecular descriptor called molecular signature was used to build a predictive model for fragrance. However, the different molecular characteristics cannot be covered using a single descriptor. Moreover, the presence or absence of certain molecular signatures was used in building the predictive model. The shortcoming of such an approach was that typical databases contain different types of molecules with very few common signatures appearing in the different molecules. Therefore, the model had to be developed using a very small subset of the database. While this approach can develop models with a low number of false positives, it leads to a high percentage of false negatives. Finally, the dilution of the fragrance molecule was not incorporated in the development of the model. However, from the fragrance molecule database, it is clear that the same molecules possess different fragrance characteristics at different concentrations. To address the limitations of the previous RSML approach, there is a need for a model that makes use of various molecular descriptors that consider a variety of structural characteristics and also the ability to make use of the available data. The approach developed in this model has attempted to address these research gaps.
To conclude, CAMD is an important approach to expanding the portfolio of chemical product design. Prediction models for scent and physical properties must be available so that the desired attributes can be incorporated as constraints in CAMD. However, due to the lack of established mechanistic odour predictive models, it is necessary to develop an empirical model for aroma using ML. This approach can generate models from data by detecting and summarising the underlying patterns. The potential of ML to generate odour predictive models can address the inherent lack of understanding of the olfaction process.
1.2. Topological Indices (TIs)
In general, the models of group contribution (GC) are extensively applied to describe the pure component properties based on molecular structure. However, differentiation of molecule position in a compound cannot be achieved by the additive group contribution methods. Even a small distinction of group position in isomers might affect the odour characteristic of molecules [
17]. Since fragrances are made up of multiple building blocks, there should be other structural attributes that contribute to fragrance in addition to the groups [
3]. Thus, topological indices, the most used descriptors for chemical structure, have been used in this study to relate molecular structure to their fragrance.
Topological indices (TIs) are molecular structure descriptors that are generated from a chemical molecular graph that characterises its topology. There are a huge number of topological indices, which can be further categorised into a few groups such as degree, spectrum and distance [
18]. Representing the chemical species using Tis provides convenience as they encode the topological structure into a mathematical form. TIs are applied extensively in developing QSPRs, which are mathematical correlations between molecular structures and molecular properties [
19]. For instance, TIs were utilised in QSPR modelling to predict the biodegradability of the molecules for the development of safer fragrance molecules [
20]. The results have shown that there are two remarkable TIs that contribute to the biodegradability of the molecules studied.
In a related study, De Mello Castanho Amboni et al. [
21] explained that the structural parameters, including TIs, are related to the odour of aliphatic esters. From the QSAR study, it is notable that the TIs such as the electro topological state index and second order shape index, Kappa 2, are the relevant molecular descriptors for odour prediction. Nevertheless, the study conducted by Chacko et al. [
22] has shown that the third-order shape index, Kappa 3, is one of the most crucial TIs for the categorisation of distinct odours. From the study by Ham and Jurs [
23], the first-order chi connectivity index and molar refractivity are the distinguishing characteristics of musk and non-musks. Therefore, several TIs are used in this study for the development of odour-predictive models as they can shed light on the structure-odour relationship of fragrance molecules. Since there are no comprehensive predictive models for fragrance prediction, machine learning approaches have been explored to relate topological indices to olfaction.
1.3. Rough Set-Based Machine Learning (RSML)
ML is a subset of artificial intelligence (AI) and consists of techniques to discover patterns in data, which can then be used for future prediction or other related tasks [
24]. Artificial neural networks (ANNs) and support-vector machines (SVMs) are particularly versatile and popular supervised ML techniques [
25]. Despite the extensive applications of SVM and ANN in QSPR, QSAR, and GC modelling, their black box nature is a crucial weakness. The outputs of ANN and SVM cannot be translated into insights easily, making it difficult to support the decision provided by the algorithms [
26]. This lack of inherent interpretability can only be addressed using additional algorithms [
27]. One alternative approach is the utilisation of inherently interpretable models [
28]. For example, hyperbox and RSML techniques can generate rule-based predictive models that are directly interpretable because they readily map to human thought processes. Because of this feature, they are better alternatives for the prediction of olfaction characteristics. Hyperbox ML has significant potential due to its ability to provide intuitive prediction accuracy in the identification of disjoint data regions [
29]. However, there are computational challenges with large datasets with imperfections (e.g., non-deterministic patterns). On the other hand, RSML has advantages for the determination of more odour characteristics. RSML has proven to be especially robust for dealing with vagueness, imprecision, inconsistency and uncertainty in datasets [
30].
Rough set theory (RST) which was first introduced by Pawlak [
31], possesses the rough equality key concept for the designated sets in a given space. An approximation space is considered a pair
, where
is a certain set known as the universe and
is an indiscernibility relation [
31]. In RST, any vague concept will be substituted by a pair of precise concepts, which is known as the lower and upper approximation of the vague concept [
32]. The major advantage of utilising RST is that there is no preliminary or additional information required regarding the data [
33]. RST has been applied in the areas of decision making, pattern recognition and knowledge acquisition. The very few early applications of rough set theory are mainly in the medical field for clinical data reduction applications and decision-making scenarios [
34], rough classification of highly selective vagotomy (HSV) patients [
35], reduction in information systems for medical diagnosis [
36], etc. Recently, RSML has been employed to determine secure geological reservoirs to minimize the unintended release of CO
2 by analysing data from secure and insecure storage sites of CO
2. The results showed the prediction models generated from RSML are comparable with the site selection rules that were constructed based on proficient knowledge [
37]. In addition, the RST was utilised as the front-end processor for deep learning to reduce the redundant influencing factors and to identify the critical factors of building energy consumption [
38].
The key concept of RST is its indiscernibility relation, which could be tabulated into an information table. It is also known as an information system or attribute-value table, which consists of objects and their corresponding attributes [
32]. The latter is comprised of conditional attributes (inputs) and decision attributes or classes of the object (outputs). An information system is defined by a pair
, where
is the finite nonempty set of objects (universe) and
is the objects ‘attributes. For every attribute
, it has a value set defined by a value,
as shown in Equations (1) and (2) [
39].
where
is the set of conditional attributes, and
is the decision attribute.
Furthermore, RST also enables the identification of reducts, defined as a minimal subset of attributes that preserve the indiscernibility relation. In the context of RSML, a reduct is a reduced set of attributes that can be used to generate a rule-based model. It should be noted that there may be more than one reduct set in a single dataset. Therefore, further analysis is required to determine which reduct can generate more feasible rules. Another important concept in RST is the intersection of all reducts, which is known as the core. It is the most important subset of attributes that contribute to classification accuracy [
32].
For every information system, there is a set of decision rules known as a decision algorithm. Each decision algorithm reveals certain properties that fulfil both the total probability theorem and Bayes’ theorem [
40]. Hence, these properties provide a new method for concluding the data by using three terms, namely strength
, certainty
and coverage
, as presented in Equations (3)–(5). Let
, where
is the conditions and
is the decisions [
33].
where
and
.
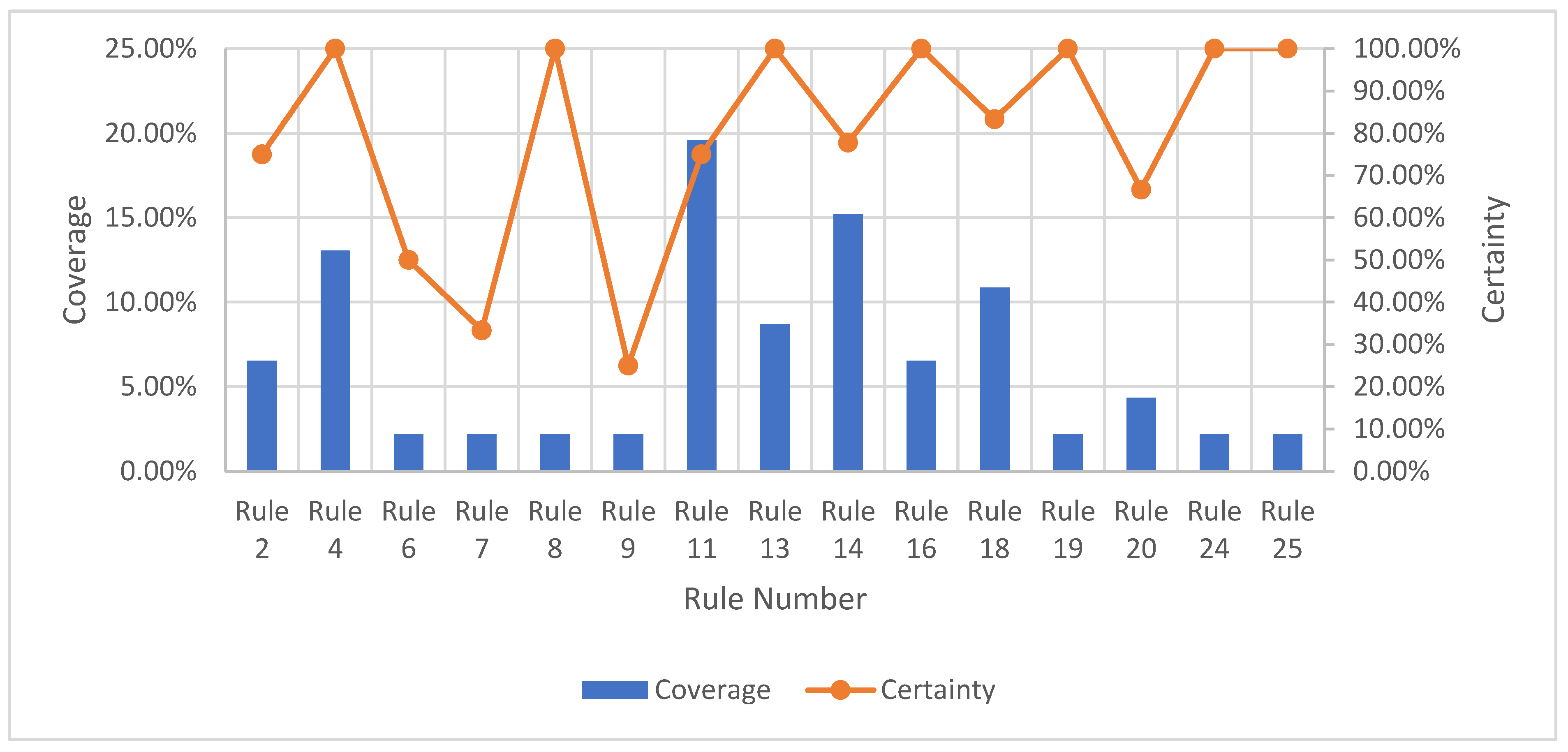
The strength represents the total number of samples that follow the generated rule divided by the total number of samples. The certainty factor is defined as the frequency of samples having the decision, , in the sets of samples that fulfil conditions, . Lastly, the coverage factor is the frequency of samples possessing conditions, in the decision class. The former measures the predictive reliability of a rule, whilst the latter measures the generalisation power of a rule. A higher certainty indicates a lower chance of a molecule being misclassified, whereas a high coverage suggests that a rule is a good approximation of an underlying general principle. These three parameters will provide quantitative evidence to help select the most useful rule-based models.
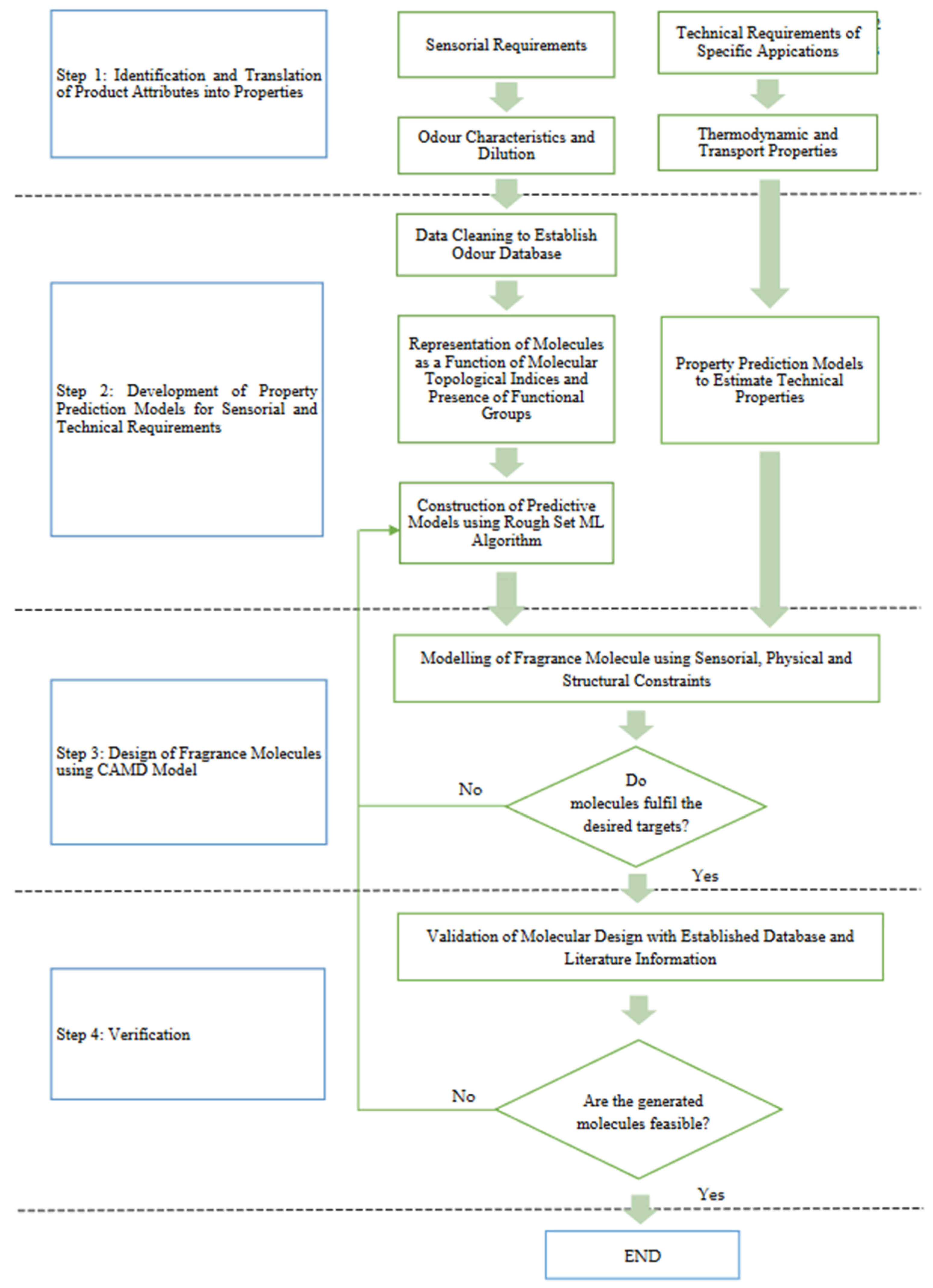
In this work, a predictive model for olfaction has been developed through RSML using structural attributes and dilution in conditional attributes. Subsequently, the most promising deterministic rules generated from RSML were integrated as constraints into CAMD, along with the structural and physicochemical constraints. For the physical properties, such as the solubility parameter and LC50, property classification is carried out, as their impacts towards the functionality of fragrances are significant only when across ranges. It is to be noted that many of the target properties are not continuous in nature. For example, the impact of toxicity and volatility does not change continuously. Therefore, the decisions on these attributes have to be measured based on the classification of toxicity or volatility classes. Current CAMD approaches only treat the properties that are continuous in nature. Therefore, disjunctive programming has been used to treat the properties where the changes over property ranges are significant. The fragrance molecule design using CAMD is formulated as a multi-objective optimisation (MOO) problem and solved using the fuzzy optimisation approach.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}