Abstract
Continuous SiC fiber is a kind of high–performance ceramic fiber that combines many advantages, such as high strength, high modulus, high hardness and low density. It has excellent mechanical properties, high–temperature and oxidation resistance, which could be applied as essential reinforcement in advanced ceramic matrix composites (CMCs) in the fields of aerospace and advanced weaponry. Melt–spinnable polytitanocarbosilane (PTCS) is an important precursor, which can be used to prepare continuous SiC fibers through a precursor–derived method. In this work, low–softening–point polycarbosilane (LPCS) and tetrabutyl titanate were used to prepare polytitanocarbosilane with a ceramic yield of 67.5 wt% at 1000 °C. FT–IR, TGA, GPC, 1H NMR, 29Si NMR, and elemental analysis were used to analyze the composition and structure of the PTCS precursor. Finally, Si–C–Ti–B fibers with an average tensile strength of 1.93 GPa were successfully prepared by melt spinning, pre–oxidation, pyrolysis, and high–temperature sintering of the PTCS precursor. The strength retention rates were 71.5% and 79.8% after heat treatment at 1900 °C and 2000 °C under an argon atmosphere for 1 h, respectively. The strength retention rates of Si–C–Ti–B fibers are higher than those of commercial Hi–Nicalon fibers, Tyranno ZMI fibers and Hi–Nicalon S fibers. This work can lay a theoretical foundation and technical support for developing high-performance SiC ceramic fibers containing titanium and their ceramic matrix composites.
1. Introduction
With high tensile strength, high elastic modulus, excellent thermal stability, good oxidation resistance and corrosion resistance, silicon carbide fibers are one of the most important reinforcements in advanced composite materials for applications in the fields of aircraft engine, spacecraft and nuclear industry [1,2,3,4,5]. The two mainstream methods currently used to prepare SiC fibers are chemical vapor deposition (CVD) and precursor-derived method. The preparation of SiC fibers by chemical vapor deposition is achieved by depositing SiC on a core material through a gas-phase reaction. C fiber and W fiber are commonly used for core materials [6,7]. The preparation cost of SiC fibers using this method is high, although the mechanical properties of the fibers are good. In addition, it is usually several times larger in diameter than SiC fibers prepared by the precursor-derived method. For example, SCS series fibers have diameters greater than 70 μm, which makes it challenging for them to be weaved into fabrics for application [8]. The precursor-derived method for preparing continuous SiC fibers with fine diameter and excellent weavability is efficient, low-cost and suitable for scale up production [9]. Since Yajima and coworkers first synthesized precursor polycarbosilane (PCS) from polydimethylsiloxane (PDMS) and successfully prepared SiC fibers using the precursor [10,11], the high temperature stability and oxidation resistance of SiC fibers have been continuously improved by the optimization of their elemental composition and microstructure. Thus far, three generations of SiC fibers have been commercialized [12,13,14,15,16,17,18,19,20,21,22,23,24,25].
SiC fibers prepared with polycarbonsilane doped with metal elements have better resistance to high temperature and oxidation than SiC fibers prepared with polycarbonsilane not doped with metal elements, while the doping of metal elements can improve the ceramic yield of polycarbonsilane [26]. It is well known that increasing the ceramic yield of polycarbosilane can optimize the properties of SiC fibers. People have been studying polytitanocarbosilane since the 1980s. PTCS were mainly synthesized by the reaction of alkoxy titanium compounds with polysilane or polycarbosilane [27]. For example, PTCS were synthesized through the reaction of PDMS and tetrabutyl titanate with the presence of polyborodiphenylsiloxane (PBSO) [26]. Song and coworkers synthesized the PTCS precursor with higher titanium content and spinnability, and successfully prepared SiC fibers containing titanium with a tensile strength of 1.2 GPa [28]. Yang et al. used small molecule silane, tetrabutyl titanate, and PBSO to prepare PTCS with a ceramic yield of 84 wt% and subsequently prepared SiC fibers with a tensile strength of 0.91 GPa [29]. Feng et al. synthesized PTCS with different titanium contents by using low molecular weight polysilane (LPS) and tetrabutyl titanate [30]. Wang and coworkers synthesized PTCS with different carbon contents by adding polyvinylchloride (PVC) and prepared carbon–rich titanium–containing SiC fibers with adjustable resistivity and tensile strength of up to 1.79 GPa [31]. Peng and Hwang improved the method of Amoros [32] to synthesize Ti–containing polycarboxymethylsilanes with linear chain structure, narrow dispersion, and a ceramic yield of 64 wt% [33]. Yu and coworkers prepared hyperbranched polytitanocarbosilane [34,35], which was mainly used for the preparation of composite materials. Tang et al. prepared a dark blue polytitanocarbosilane precursor using PDMS and tetrabutyl titanate, which was then used as the precursor to prepare a SiO2/TiO2 fiber with strength varying with the preparation temperature [36]. He and Zhang et al. used liquid polysilane, tetrabutyl titanate, tetrabutyl borate, and solid polysilane with a softening point at 198 °C to prepare a boron–modified polytitanocarbosilane precursor, and then prepared SiC fibers containing Ti and B elements with tensile strength ranging from 0.76 to 1.36 GPa [37]. In addition to the introduction of Ti to polycarbosilanes, some researchers have added Zr to polycarbosilanes to improve the ceramic yield of polycarbosilanes.Vijay et al. synthesized Polyzirconiumcarbosilane using Zr(acac)4 and PCS with a ceramic yield of up to 76 wt%, increasing the ceramic yield of the PCS by 39 wt% [38]. Polyzirconiumcarbosilane was prepared using bis(cyclopentadienyl) zirconium dichloride, vinylmagnesium chloride, and LPCS with a ceramic yield of 73.6 wt% by Chen et al. [39]. Additionally, Gou et al. prepared polyaluminocarbosilane by the reaction of LPCS and aluminum acetylacetonate with a ceramic yield of 60 wt% [40].
Many other metal elements can be doped, and of course, many researchers have improved the ceramic yield of polycarbosilane by other methods. Su et al., prepared a high–viscosity polycarbosilane with a ceramic yield of 77 wt% using liquid polycarbosilane, Cl2CHSiMeCl2, and CH2 = CHCHCH2Cl as reaction materials [41]. Wang et al., prepared a light-sensitive acrylate-grafted PCS that can be cured by UV irradiation when using this PCS for fiber preparation [42]. Chen et al. prepared a cyano–polycarbosilane by introducing cyano into PCS, and the ceramic yield of this precursor was 80 wt% [43]. Wang et al. designed and synthesized vinyl ether grafted liquid polycarbosilane (VE–LHBPCS) with the approximate formula [SiH1.95(OCH2CH2OCH = CH2)0.05CH2]n and the ceramic yield of this precursor was 70 wt% [44]. Yao et al., synthesized vinyl–modified polycarbosilane using PCS and DVS catalyzed by Karstedt’s catalyst, and the ceramic yield of this precursor was 77.6 wt%, which was 9.3 wt% higher than that of the raw material PCS [45].
Previous studies synthesized PTCS precursors with complicated processes, and the strength retention rate of the SiC fibers obtained from the preparation was low. In this work, for the first time, PTCS precursors with good spinning performance were synthesized from low-softening-point polycarbosilane (LPCS). The overall process was simple and easy to operate. Furthermore, the as-synthesized PTCS was used to successfully prepare SiC fibers with good mechanical properties and excellent high–temperature resistance.
2. Experiment
2.1. Materials
The low-softening-point polycarbosilane (LPCS) was synthesized by our laboratory, and has a softening point of 170 °C. Tetrabutyl titanate (Ti(OCH2CH2CH2CH3)4) and xylene were purchased from Inno-chem. All other reagents and solvents were obtained in the highest purity and used without further purification unless otherwise stated.
2.2. Characterization
The softening point of LPCS was tested using a Model 9200 melting point tester with a heating rate of 1 °C/min and a test temperature from room temperature to 220 °C. Three samples were placed at a time, and the operation was repeated three times to obtain the average value. The molecular weight and dispersion of the polymers were measured by a Wyatt Gel Permeation Chromatography (GPC) with tetrahydrofuran used as the eluent at a flow rate of 1 mL/min and operated at 30 °C. The polymers were analyzed for their chemical structure and composition by Bruker Fourier Transform Infrared Spectroscopy with the measurement range from 400 to 4000 cm−1. 1H NMR and 29Si NMR spectra of the polymers were obtained on a Bruker Avance NEO 600 spectrometer at 600 MHz, and all chemical shifts are reported in ppm with reference to chemical shifts of solvent resonances. Thermal Gravimetric Analysis (TGA) was performed using a STA2500 Regulus thermogravimetric analyzer (Perkin Elmer, Waltham, MA, USA) with a heating range of room temperature to 1000 °C, a heating rate of 10 °C/min, and a nitrogen flow rate of 50 mL/min. The calculation method of the ceramic yield in this article is based on the results of thermogravimetric analysis to retrieve the mass retention rate of the PTCS precursor at 1000 °C. The elemental contents of Ti was measured by chemical analysis. The SiC fiber morphology was studied by Field Emission Scanning Electron Microscopy Tescan Mira3. The tensile strength and modulus of SiC fibers were measured by using an Instron tester (Testometric, M350-5CT, Boston, MA, USA) with a gauge length of 25 mm. The measurement details are as follows: Prepare a coordinate paper with several squares, each with a side length of 25 mm. Cut out these squares to obtain a coordinate paper with several hollow cells. Fix a single fiber (length > 25 mm) onto the hollow cell using adhesive to have an exposed fiber length of 25 mm. Then, cut the coordinate paper to an appropriate size before transferring it to the testing equipment for measurement. The average tensile strength was obtained from the measurements of 25 monofilaments.
2.3. Synthesis of Polytitanocarbosilane
LPCS and Ti(OCH2CH2CH2CH3)4 were dissolved in xylene and the reaction mixture was heated to over 200 °C for the chemical reaction. After being cooled to ambient temperature, the reaction mixture was filtered. Then, it was distilled under vacuum to obtain the PTCS precursor. PTCS was a black glossy solid with a ceramic yield of 67.5 wt%.
2.4. Preparation of Si–C–Ti–B Fibers
The PTCS precursor was heated to above 300 °C for melt spinning to prepare green fibers. Green fibers were then heated to about 200 °C at a heating rate of 9 °C/h and held for 2 h for oxygen-curing to obtain pre-oxidized fibers. PTCS molecules in green fibers were further cross-linked during this process to convert green fibers from thermoplastic to thermosetting, and to prevent green fibers from melting and sticking together in the subsequent heat treatment process. Pre-oxidized fibers were subsequently heated to over 1000 °C at a heating rate of 117 °C/h under B–containing atmosphere to obtain Si–C–O–Ti–B fibers, which were finally heated to 1800 °C at a heating rate of 7 °C/min and held at 1800 °C to obtain Si–C–Ti–B fibers, during which the SiCxOy phase in the Si–C–O–Ti–B fibers was decomposed, releasing CO and SiO gases, and the β–SiC in Si–C–O–Ti–B fibers further crystallized. In this process, Si–C–O–Ti–B fibers were sintered and densified.
2.5. High–Temperature Resistance Testing of Si–C–Ti–B Fibers
The Si–C–Ti–B fibers were heated under an argon atmosphere from room temperature to 1800 °C, 1900 °C, and 2000 °C, respectively, and held at the target temperature for 1 h, then were cooled in the furnace. The heating rate from room temperature to 1000 °C was 10 °C/min, and the heating rate over 1000 °C was 5 °C/min.
3. Results and Discussion
3.1. Synthesis and Characterization of the PTCS Precursor
In this work, the PTCS precursor is synthesized by the reaction of LPCS with Ti(OCH2CH2CH2CH3)4 at a temperature above 200 °C. The mass ratio of Ti(OCH2CH2CH2CH3)4 to LPCS is 1:10, and PTCS with good melt spinning properties is obtained. The Ti content in the PTCS precursor is measured as 1.43 wt%. An optical photo of PTCS is shown in Figure 1a. It can be seen that PTCS is a black glossy solid. In order to qualitatively analyze the molecular structure of the PTCS precursor, FT–IR characterization is performed, and the result is shown in Figure 1b. The stretching vibration peaks of the C–H bonds are located at 2950 cm−1, Si–C bonds at 820 cm−1, and Si–CH2–Si bonds at 1030 cm−1. Furthermore, it is also proved the presence of the peaks of the Si–H bonds (2100 cm−1, the stretching vibration), Si–CH3 bonds (1250 cm−1, stretching vibration), and Ti–O–R bonds (stretching vibration peak, 400–950 cm−1) formed by the condensation reaction between the active Si–H bonds with Ti(OCH2CH2CH2CH3)4. In addition, it can be seen that there are remaining Si–H bonds in the PTCS, which mean that more Ti elements could be introduced into the PTCS precursor if needed by the method described herein.

Figure 1.
(a) Optical photo of the PTCS precursor. (b) Infrared spectrum of LPCS and PTCS.
The 1H NMR spectrum of precursors in Figure 2a shows the peaks with chemical shifts around −0.30 ppm, 0 ppm, 0.18 ppm, 0.92 ppm, 1.36 ppm and 4.65 ppm, which prove the presence of CH2, Si(CH3)4, Si–CH3, O–alkyl, –OH and Si–H groups in the PTCS precursor. In addition, the peaks around 4.65 ppm also confirm the presence of remaining Si–H bonds in the PTCS precursor. This is consistent with the results of the FT–IR spectrum. From 29SiNMR spectrum of precursors in Figure 2b, it can be seen that there are two peaks located around −16 and 0 ppm, which could be attributed to SiC3H and SiC4 bonds, respectively. The content ratio of SiC3H to SiC4 bonds of LPCS and the PTCS precursor could be calculated by comparing the corresponding areas of the two peaks, which are about 1.64 and 1.39, respectively.

Figure 2.
NMR spectra of LPCS and PTCS: (a) 1H NMR; (b) 29Si NMR.
It can be seen from Figure 3a that the GPC curves of LPCS and PTCS exhibit a broad peak. The number-average molecular weights of LPCS and PTCS are about 1126 and 1140, and the weight-average molecular weights are about 2619 and 2516, respectively. Hence, the polydispersity indexes are 2.32 and 2.21, respectively. Figure 3b shows the thermogravimetric analysis of the polymer heated to 1000 °C at 10 °C/min under nitrogen. The weight loss process of the PTCS precursor has three main stages as follows: the weight loss of the PTCS precursor below 300 °C is mainly caused by the escape of the low-molecular-weight molecules in the PTCS precursor into gaseous state; the weight loss of the PTCS precursor from 300 °C to 500 °C consists of two parts, one is that the low-molecular-weight molecules in PTCS precursor continue to be heated to gaseous state, and the other is that the remaining Si–H bonds and Si–CH3 in the PTCS precursor further experience dehydrogenation crosslinking, and the release of H2 accompanies the process; the weight loss of the PTCS precursor from 500 °C to 800 °C is mainly due to the thermal decomposition of Si–H, Si–CH3, and other side groups of the PTCS precursor. The rapid weight loss of the PTCS precursor is caused by releasing a large number of small molecules of hydrocarbons such as CH4 and H2. The transformation of the PTCS precursor from organic to inorganic happens at this stage, which is also named as the ceramicization of the PTCS precursor. The weight loss of the PTCS precursor below 300 °C is negligible. The PTCS precursor starts to lose weight significantly at 339 °C and shows a rapid weight loss from 350 °C to 550 °C. The ceramization is basically completed at about 800 °C. The ceramic yields at 1000 °C for LPCS and PTCS were 49.9 wt% and 67.5 wt%, respectively. Compared with LPCS, PTCS has a 17.6 wt% higher ceramic yield, which is high enough for it being used as the precursor of SiC ceramic fibers. The PTCS precursor had a 7.5 wt% higher ceramic yield than the PACS precursor prepared by Gou et al. using LPCS and aluminum acetylacetonate [40].
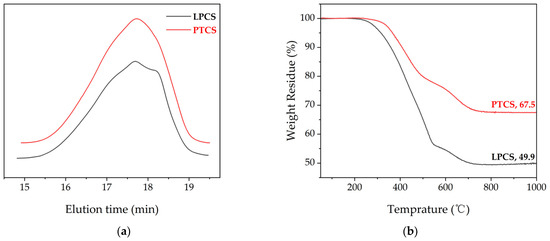
Figure 3.
(a) GPC curves of LPCS and PTCS. (b) TG curves of LPCS and PTCS.
3.2. Preparation and Characterization of the Si–C–Ti–B Fibers Derived from PTCS
The aforementioned PTCS precursor is used to prepare Si–C–Ti–B fibers through sequential processes of melt spinning, pre-oxidation, pyrolysis and high-temperature sintering. The green fibers were pre-oxidized to obtain pre–oxidized fibers. The ceramic yield of pre–oxidized fibers at 1000 °C is 83.3 wt%, which is comparable to that of the preoxidized fibers prepared by He et al. [37]. Figure 4 shows the SEM images of the Si–C–O–Ti–B fibers. From Figure 4a, it can be seen that the fiber surface is smooth and dense, and further magnification reveals the presence of numerous small cracks on the fiber surface. Figure 4c,d show that the core of the Si–C–O–Ti–B fibers cross–section is relatively loose and contains a small number of micropores, while the areas closer to the edge of the fiber cross–section are denser. In addition, it can be observed that there is a less–apparent carbon–rich layer on the surface of the Si–C–O–Ti–B fibers. From Figure 5, it can be seen that the diffraction peaks of Si–C–O–Ti–B fibers are not very sharp, indicating that the Si–C–O–Ti–B fibers are not completely crystallized and still retain some amorphous phases. The characteristic diffraction peaks of the (110) crystal plane of SiO2 appear near 2θ = 20.9°, and the (111), (220) and (311) crystal plane of β–SiC appear near 2θ = 35.4°, 60.3° and 71.7°, respectively. In addition, the average tensile strength of the fiber is 1.72 GPa.

Figure 4.
SEM images of the Si–C–O–Ti–B fibers. (a) Surface of Si–C–O–Ti–B fibers. (b) Enlarged view of the surface morphology. (c) Cross–section of Si–C–O–Ti–B fibers. (d) Enlarged view of the cross section morphology.
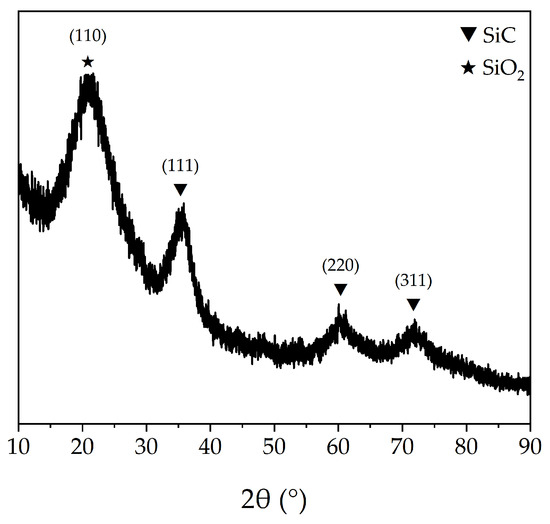
Figure 5.
XRD pattern of Si–C–O–Ti–B fibers.
Figure 6 shows the SEM images of Si–C–Ti–B fibers obtained at 1800 °C. From Figure 6a,b, it can be seen that the overall surface morphology of the Si–C–Ti–B fiber is rough, with an average diameter of 13.1 μm. A large number of carbon–rich particles with sizes ranging from tens to hundreds of nanometers can be observed on the surface of Si–C–Ti–B fibers. The cross-section of the Si–C–Ti–B fiber is shown in Figure 6c,d, and there are basically no pores in the fiber core. The Si–C–Ti–B fibers obtained at 1800 °C become denser due to the high-temperature sintering. In addition, compared with Si–C–O–Ti–B fibers, the interface between the coating on the Si–C–Ti–B fiber surface and the fiber core becomes clearer, and an apparent carbon–rich layer with a thickness of 463.2 nm can be observed at the edge of the fiber cross–section. From the XRD results of Si–C–Ti–B fibers in Figure 7, it can be seen that the overall crystallinity of the fibers is much higher than the Si–C–O–Ti–B fibers. The eight peaks belonging to TiB2 correspond to (001), (100), (101), (002), (110), (102), (111), and (201) crystal planes, while the remaining five peaks correspond to the (111), (200), (220), (311), and (222) crystal planes of β–SiC. In addition, the average grain size of β–SiC in the Si–C–Ti–B fibers is calculated by Scheller formula as 39.3 nm. It is noteworthy that the peaks of TiB2 could also be observed clearly, which confirms the presence of TiB2 grains. The average tensile strength of the Si–C–Ti–B fibers was 1.93 GPa, which was 0.21 GPa higher than that of the Si–C–O–Ti–B fibers.

Figure 6.
SEM images of the Si–C–Ti–B fibers obtained at 1800 °C. (a) Surface of Si–C–Ti–B fibers. (b) Enlarged view of the surface morphology. (c) Cross–section of Si–C–Ti–B fibers. (d) Enlarged view of the cross section morphology.

Figure 7.
XRD pattern of Si–C–Ti–B fibers obtained at 1800 °C.
In order to investigate the high–temperature resistance of as prepared Si–C–Ti–B fibers, the Si–C–Ti–B fibers were heated to 1900 °C, 2000 °C, and 2100 °C under argon atmosphere and held for 1 h. The SEM images of the Si–C–Ti–B fibers after heat treatment are shown in Figure 8. The surfaces of Si–C–Ti–B fibers were smooth and dense after heat treatment at different temperatures. However, the decomposition of SiC occurs in the interface region between the carbon-rich layer and the fiber core. With the increment of heat treatment temperature, the SiC grains in Si–C–Ti–B fiber decompose more obviously. After the high-temperature treatment at 2100 °C, the decomposition of SiC and the growth of SiC grains can be significantly observed, which is similar to the literature [46]. During this process, the decomposition of Si–C–Ti–B fibers gradually happens from the carbon–rich layer to the fiber core, resulting in a porous cross–section. It is noteworthy that the core of Si–C–Ti–B fibers remains dense. The tensile strengths of Si–C–Ti–B fibers were 1.38 GPa and 1.54 GPa after heat treatment at 1900 °C and 2000 °C, respectively, with tensile strength retention rates of 71.5% and 79.8%, respectively. One of the possible reasons for the superior mechanical properties of Si–C–Ti–B fibers after heat treatment at 2000 °C compared to those after heat treatment at 1900 °C is that Si–C–Ti–B fibers have a more dense core after heat treatment at 2000 °C. The strength of Si–C–Ti–B fibers decreased so significantly after heat treatment at 2100 °C that it could not be measured.

Figure 8.
SEM images of the surface (a,c,e) and cross–section (b,d,f) of the Si–C–Ti–B fiber after heat treatment for 1 h under argon atmosphere at 1900 °C (a,b), 2000 °C (c,d), and 2100 °C (e,f), respectively.
The XRD results of Si–C–Ti–B fibers after heat treatment at different temperatures are shown in Figure 9. Compared with Si–C–Ti–B fibers without heat treatment, TiC phases appear in Si–C–Ti–B fibers after heat treatment at 1900 °C, 2000 °C, and 2100 °C, formed by the reaction between Ti and C. A C(002) diffraction peak can be observed for Si–C–Ti–B fibers after heat treatment at 2100 °C. This result corresponds to the significant decomposition of SiC grains in Figure 8f. Interestingly, the TiB2 phase is also observed for Si–C–Ti–B fibers after heat treatment at 2100 °C. The diffraction peaks of (110) and (101) crystal planes of TiB2 can be seen in Figure 9. The average sizes of β–SiC grains in Si–C–Ti–B fibers after heat treatment at 1900 °C, 2000 °C and 2100 °C were calculated to be 42.6 nm, 43.5 nm, and 50.6 nm, respectively. It can be seen that the β–SiC grain size in Si–C–Ti–B fibers rapidly increases when the heat treatment temperature increases to 2100 °C, which proves the growth of the remaining SiC that has not yet decomposed.

Figure 9.
XRD pattern of Si–C–Ti–B fibers after heat treatment at 1900 °C, 2000 °C and 2100 °C under argon atmosphere for 1 h.
The strength retention rate of Si–C–Ti–B fibers after heat treatment at different temperatures under an inert atmosphere for 1 h is compared with that of several typical commercial SiC fibers, as shown in Figure 10 [2,3,47]. Hi–Nicalon fibers, Tyranno ZMI fibers, and F–2 fibers are typical 2nd generation SiC fibers, and Hi–Nicalon S fibers and F–3 fibers are typical 3rd generation SiC fibers. It can be seen from Figure 10 that the strength retention rates of Si–C–Ti–B fibers are higher than those of Hi–Nicalon fibers, Tyranno ZMI fibers, F–2 fibers, and Hi–Nicalon S fibers.

Figure 10.
Strength retention rate of Si–C–Ti–B fibers and some commercial SiC fibers after heat treatment at different temperatures under inert atmosphere for 1 h.
4. Conclusions
In this work, we synthesized PTCS precursors using low–softening–point polycarbonsilane (LPCS) and Ti(OCH2CH2CH2CH3)4 as raw materials for the first time, which significantly shortened the reaction process compared to previous studies. The ceramic yield of the PTCS precursor was 67.5 wt%, which is comparable to the ceramic yield of other polycarbonsilane precursors for melt spinning. The synthesized PTCS was proved as an excellent precursor for melt spinning to prepare SiC fibers. The overall procedure is simple and easy to implement, and the method can be further extended to synthesize other heterogeneous metal–containing polycarbosilane precursors. Subsequently, melt spinning of the PTCS precursor was carried out to obtain green fibers, which were then pre–oxidized, pyrolysis, and high–temperature sintered to obtain Si–C–Ti–B fibers with an average tensile strength of 1.93 GPa. High–temperature resistance tests were also performed. The tensile strengths of Si–C–Ti–B fibers were 1.38 GPa and 1.54 GPa, and the tensile strength retention rates were 71.5% and 79.8% after treatment at 1900 °C and 2000 °C under argon atmosphere for 1 h, respectively. The strength retention rates of Si–C–Ti–B fibers were higher than those of commercial Hi–Nicalon fibers, Tyranno ZMI fibers and Hi–Nicalon S fibers. The results proved that Si–C–Ti–B fibers have excellent high–temperature resistance. The PTCS precursor can be used not only for preparing continuous ultra–high temperature resistant SiC ceramic fibers, but also for preparing SiC ceramic matrix composites.
Author Contributions
Conceptualization, Y.G.; methodology, Y.G.; validation, Q.Z., T.C. and W.K.; formal analysis, Q.Z. and S.W.; investigation, Q.Z.; writing—original draft preparation, Q.Z.; writing—review and editing, Y.G. and X.X.; supervision, Y.G.; project administration, Y.G. All authors have read and agreed to the published version of the manuscript.
Funding
The financial support from National Natural Science Foundation of China (Grant No. 51772327), Natural Science Foundation of Hunan Province (Grant No. 2022JJ30662), Science and Technology on Advanced Ceramic Fibers and Composites Laboratory (Grant No. WDZC20205500504, WDZC20215250507) and National Key R&D Program of China (2022YFB3707700) are greatly appreciated.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, P.; Liu, F.; Wang, H.; Li, H.; Gou, Y. A Review of Third Generation SiC Fibers and SiCf/SiC Composites. J. Mater. Sci. Technol. 2019, 35, 2743–2750. [Google Scholar] [CrossRef]
- Wang, P.; Gou, Y.; Wang, H. Third Generation SiC Fibers for Nuclear Applications. J. Inorg. Mater. 2020, 35, 525. [Google Scholar] [CrossRef]
- Gou, Y.; Wang, H.; Jian, K. Formation of Carbon–Rich Layer on the Surface of SiC Fiber by Sintering under Vacuum for Superior Mechanical and Thermal Properties. J. Eur. Ceram. Soc. 2017, 37, 907–914. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, S.; He, A. Application Requirements and Challenges of CMC–SiC Composites on Aero–Engine. J. Mater. Eng. 2019, 47, 1–10. [Google Scholar] [CrossRef]
- Yin, X.W.; Cheng, L.F.; Zhang, L.T.; Travitzky, N.; Greil, P. Fibre–Reinforced Multifunctional SiC Matrix Composite Materials. Int. Mater. Rev. 2017, 62, 117–172. [Google Scholar] [CrossRef]
- Okada, K.; Kato, H.; Nakajima, K. Preparation of Silicon Carbide Fiber from Activated Carbon Fiber and Gaseous Silicon Monoxide. J Am. Ceram. Soc. 1994, 77, 1691–1693. [Google Scholar] [CrossRef]
- Liu, S.; Luo, X.; Huang, B.; Li, P.; Yang, Y. Role of H2 and Ar as the Diluent Gas in Continuous Hot–Wire CVD Synthesis of SiC Fiber. J. Eur. Ceram. Soc. 2022, 42, 3135–3147. [Google Scholar] [CrossRef]
- Flores, O.; Bordia, R.K.; Nestler, D.; Krenkel, W.; Motz, G. Ceramic Fibers Based on SiC and SiCN Systems: Current Research, Development, and Commercial Status. Adv. Eng. Mater. 2014, 16, 621–636. [Google Scholar] [CrossRef]
- Chu, Z.; Feng, C.; Song, Y.; Li, X.; Xiao, J.; Wang, Y. Research and Development of SiC Using the Precursor Conversion Method and Continuous Fiber Technology Both Domestically and Internationally. J. Inorg. Mater. 2002, 17, 193–201. [Google Scholar]
- Yajima, S.; Hayashi, J.; Omori, M. Continuous Silicon Carbide Fiber of High Tensile Strength. Chem. Lett. 1975, 4, 931–934. [Google Scholar] [CrossRef]
- Yajima, S.; Hayashi, J.; Omori, M.; Okamura, K. Development of a Silicon Carbide Fibre with High Tensile Strength. Nature 1976, 261, 683–685. [Google Scholar] [CrossRef]
- Bunsell, A.R.; Piant, A. A Review of the Development of Three Generations of Small Diameter Silicon Carbide Fibres. J. Mater. Sci. 2006, 41, 823–839. [Google Scholar] [CrossRef]
- Li, L.; Jian, K.; Wang, Y. Study of Oxidation Resistance of KD–I and KD–II Continuous SiC Fibers in Air. Mater. Rev. 2016, 30, 308–312. [Google Scholar]
- Vahlas, C.; Rocabois, P.; Bernard, C. Thermal Degradation Mechanisms of Nicalon Fibre:A Thermodynamic Simulation. J. Mater. Sci. 1994, 29, 5839–5846. [Google Scholar] [CrossRef]
- Vahlas, C.; Laanani, F. Thermodynamic Study of the Thermal Degradation of SiC–Based Fibres: Influence of SiC Grain Size. J. Mater. Sci. Lett. 1995, 14, 1558–1561. [Google Scholar] [CrossRef]
- Chollon, G.; Pailler, R.; Naslain, R.; Olry, P. Correlation between Microstructure and Mechanical Behaviour at High Temperatures of a SiC Fibre with a Low Oxygen Content (Hi–Nicalon). J. Mater. Sci. 1997, 32, 1133–1147. [Google Scholar] [CrossRef]
- Yun, H.M.; DiCarlo, J.A. Comparison of the Tensile, Creep, and Rupture Strength Properties of Stoichiometric SiC Fibers. Ceram. Eng. Sci. Proc. 1999, 20, 259–272. [Google Scholar]
- Hirotsu, Y.; Wakoh, K.; Suzuki, K.; Sumiyama, K.; Yamamuro, S.; Kamiyama, T.; Shibuya, M.; Yamamura, T. High–Resolution TEM Observation of β–SiC Nano–Crystallite Evolution in Si–C–Ti–O Fibers Pyrolyzed from Polytitanocarbosilane Precursor. Mater. Trans. JIM 1997, 38, 5–10. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kohtoku, Y.; Kumagawa, K.; Yamamura, T.; Nagasawa, T. High–Strength Alkali–Resistant Sintered SiC Fibre Stable to 2200 °C. Nature 1998, 391, 773–775. [Google Scholar] [CrossRef]
- Hochet, N.; Berger, M.H.; Bunsell, A.R. Microstructural Evolution of the Latest Generation of Small-diameter SiC-based Fibres Tested at High Temperatures. J. Microsc. 1997, 185, 243–258. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kohtoku, Y.; Kumagawa, K. Production Mechanism of Polyzirconocarbosilane Using Zirconium(IV)Acetylacetonate and Its Conversion of the Polymer into Inorganic Materials. J. Mater. Sci. 1998, 33, 161–166. [Google Scholar] [CrossRef]
- Shimoo, T.; Hayatsu, T.; Narisawa, M.; Takeda, M.; Ichikawa, H.; Seguchi, T.; Okamura, K. Pyrolysis of Low–Oxygen Sic Fiber Prepared by Electron–Irradiation Curing Method. J. Ceram. Soc. Jpn. 1993, 101, 1379–1383. [Google Scholar] [CrossRef]
- Sugimoto, M.; Shimoo, T.; Okamura, K.; Seguchi, T. Reaction Mechanisms of Silicon Carbide Fiber Synthesis by Heat Treatment of Polycarbosilane Fibers Cured by Radiation: II, Free Radical Reaction. J. Am. Ceram. Soc. 1995, 78, 1849–1852. [Google Scholar] [CrossRef]
- Lipowitz, J.; Rabe, J.A.; Zangvil, A. Structure and Properties of Sylramic TM Silicon Carbide Fiber—A Polycrystalline, Stoichiometric B-Sic Composition. Ceram. Eng. Sci. Proc. 1997, 18, 147–157. [Google Scholar]
- Yun, H.M.; DiCarlo, J.A.; Chen, Y.L.; Wheeler, D.R. Thermo–Mechanical Properties of Super Sylramic SiC Fibers. In Proceedings of the Mechanical Properties and Performance of Engineering Ceramics and Composites, Cocoa Beach, FL, USA, 1 January 2005. [Google Scholar]
- Song, Y.; Feng, C.; Liu, Y.; Lu, Y. Synthesis of Precursors PTC–III of SiC Fibers Containing Ti. Mater. Sci. Prog. 1992, 6, 250–255. [Google Scholar]
- Yajima, S.; Iwai, T.; Yamamura, T. Synthesis of a Polytitanocarbosilane and Its Conversion into Inorganic Compounds. J. Mater. Sci. 1981, 16, 1349–1355. [Google Scholar] [CrossRef]
- Song, Y.; Lu, Y.; Feng, C. Synthesis of Precursors of SiC Fibers Containing Ti. Mater. Sci. Prog. 1990, 4, 436–440. [Google Scholar]
- Yang, Y.; Feng, C.; Lu, Y.; Zhang, W. Preparation of Polytitanocarbosilane and Titanium-–Containing Carbon Fiber. Aerosp. Mater. Technol. 1991, 3, 20–25. [Google Scholar]
- Wang, Y.; Feng, C.; Song, Y. Preparation and Property of Si–Ti–C–O Fibers with Adjustable Electric Resistivity. Aerosp. Mater. Technol. 1999, 29, 28–31. [Google Scholar]
- Wang, Y.; Zhao, P.; Song, Y.; Feng, C. Syntheses of Precursors of Si–Ti–C–O Fibers with Rich Carbon. Aerosp. Mater. Technol. 2001, 2, 24–27. [Google Scholar]
- Amorós, P.; Beltrán, D.; Guillem, C.; Latorre, J. Synthesis and Characterization of SiC/MC/C Ceramics (M = Ti, Zr, Hf) Starting from Totally Non–Oxidic Precursors. Chem. Mater. 2002, 14, 1585–1590. [Google Scholar] [CrossRef]
- Peng, C.H.; Hwang, C.C. Synthesis and Characteristics of Polycarbomethylsilane via a One–Pot Approach. J. Mater. Res. Technol. 2020, 9, 15838–15848. [Google Scholar] [CrossRef]
- Yu, Z.; Zhan, J.; Zhou, C.; Yang, L.; Li, R.; Xia, H. Synthesis and Characterization of SiC(Ti) Ceramics Derived from a Hybrid Precursor of Titanium–Containing Polycarbosilane. J. Inorg. Organomet. Polym. 2011, 21, 412–420. [Google Scholar] [CrossRef]
- Yu, Z.; Yang, L.; Min, H.; Zhang, P.; Liu, A.; Riedel, R. High–Ceramic–Yield Precursor to SiC–Based Ceramic: A Hyperbranched Polytitaniumcarbosilane Bearing Self–Catalyzing Units. J. Eur. Ceram. Soc. 2015, 35, 851–858. [Google Scholar] [CrossRef]
- Tang, X.; Yu, Y.; Yang, D. SiO2/TiO2 Fibers from Titanium–Modified Polycarbosilane. J. Mater. Sci. 2010, 45, 2670–2674. [Google Scholar] [CrossRef]
- He, G.; Zhang, B.; Wang, B.; Xu, D.; Li, S.; Yu, Z.; Chen, J. Amorphous Fine–Diameter SiC–Based Fiber from a Boron–Modified Polytitanocarbosilane Precursor. J. Eur. Ceram. Soc. 2018, 38, 1079–1086. [Google Scholar] [CrossRef]
- Vijay, V.V.; Nair, S.G.; Sreejith, K.J.; Devasia, R. Synthesis, Ceramic Conversion and Microstructure Analysis of Zirconium Modified Polycarbosilane. J. Inorg. Organomet. Polym. 2016, 26, 302–311. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Wang, H. Synthesis, Characterization and Pyrolysis of a High Zirconium Content Zirconocene–Polycarbosilane Precursor without Zr–O Bond. Mater. Des. 2016, 90, 84–90. [Google Scholar] [CrossRef]
- Gou, Y.; Wang, H.; Jian, K.; Wang, Y.; Wang, J.; Song, Y.; Xie, Z. Facile Synthesis of Melt–Spinnable Polyaluminocarbosilane Using Low–Softening–Point Polycarbosilane for Si–C–Al–O Fibers. J. Mater. Sci. 2016, 51, 8240–8249. [Google Scholar] [CrossRef]
- Su, G.; Ban, Z.; Li, Y.; Li, W.; Zhang, Z. Preparation, Characterization, and Pyrolysis of Polycarbosilane with High Ceramic Yield and High Viscosity. Polym. Sci. Ser. B 2022, 64, 598–605. [Google Scholar] [CrossRef]
- Wang, Y.; Pei, X.; Li, H.; Xu, X.; He, L.; Huang, Z.; Huang, Q. Preparation of SiC Ceramic Fiber from a Photosensitive Polycarbosilane. Ceram. Int. 2020, 46, 28300–28307. [Google Scholar] [CrossRef]
- Chen, D.; Mo, G.; Qian, J.; He, L.; Huang, Q.; Huang, Z. Synthesis of Cyano–Polycarbosilane and Investigation of Its Pyrolysis Process. J. Eur. Ceram. Soc. 2020, 40, 5226–5237. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, J.; Pei, X.; He, L.; Huang, Z.; Huang, Q. Preparation of Liquid Polycarbosilane Containing Vinyl Ether Group and Its Rapid Curing through Thiol-ene Click Reaction. J. Am. Ceram. Soc. 2022, 105, 7122–7131. [Google Scholar] [CrossRef]
- Yao, B.; Lu, B.; Huang, Q.; Huang, Z.-R.; Yuan, Q. The Preparation of SiC Ultrafine Fibers Containing Low Amount of Oxygen by the Electrospinning and Pyrolysis of Vinyl–Modified Polycarbosilane. Ceram. Int. 2020, 46, 9894–9900. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Yan, D.; Wang, S.; Li, G.; Gou, Y. Conversion of Silicon Carbide Fibers to Continuous Graphene Fibers by Vacuum Annealing. Carbon 2021, 182, 435–444. [Google Scholar] [CrossRef]
- Gou, Y.; Jian, K.; Wang, H.; Wang, J. Fabrication of Nearly Stoichiometric Polycrystalline SiC Fibers with Excellent High–Temperature Stability up to 1900 °C. J. Am. Ceram. Soc. 2018, 101, 2050–2059. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).