
Sodium Hypochlorite Pentahydrate as a Chlorinating Reagent: Application to the Tandem Conversion of β,γ-Unsaturated Carboxylic Acids to α,β-Unsaturated Lactones

Abstract
:1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Procedure
4.2. Reaction of (E)-4-Phenylpent-3-Enoic Acid (2a)
4.3. Reaction of (E)-4-Phenylbut-3-Enoic Acid (2b)
4.4. Reaction of (E)-4-(4-Methoxyphenyl)But-3-Enoic Acid (2c)
4.5. Reaction of But-3-Enoic Acid (2d)
4.6. Reaction of (E)-Hex-3-Enoic Acid (2e)
4.7. Reaction of (E)-5-Phenylpent-3-Enoic Acid (2f)
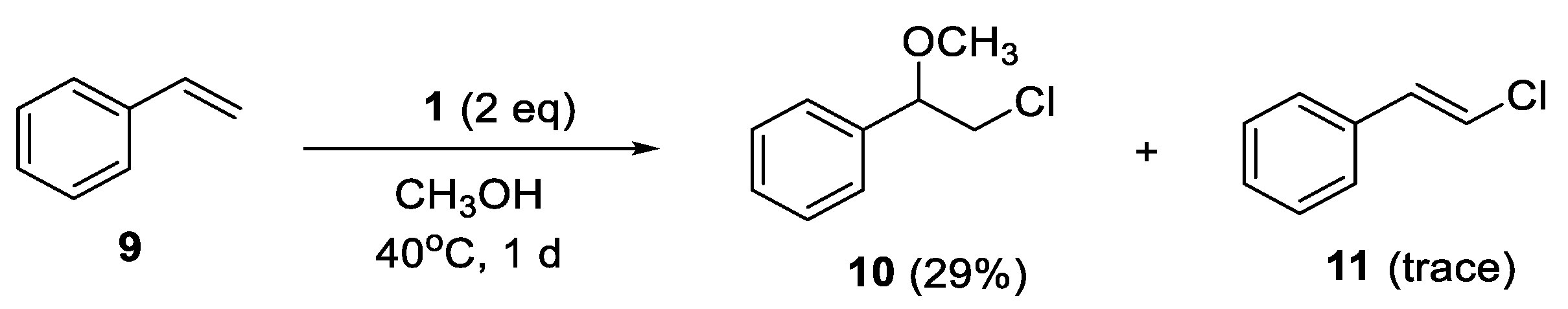
4.8. Reaction of Styrene (9)
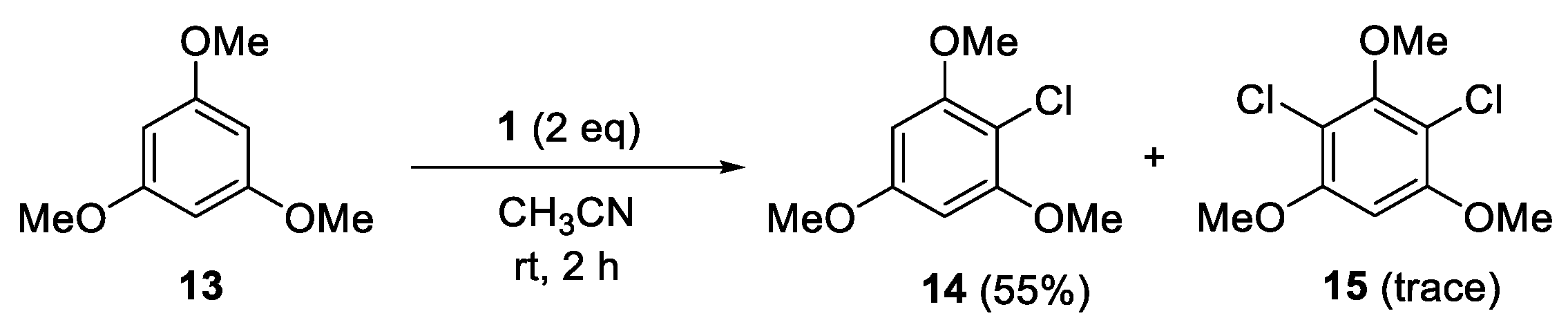
4.9. Reaction of 1,3,5-Trimethoxybenzene (13)
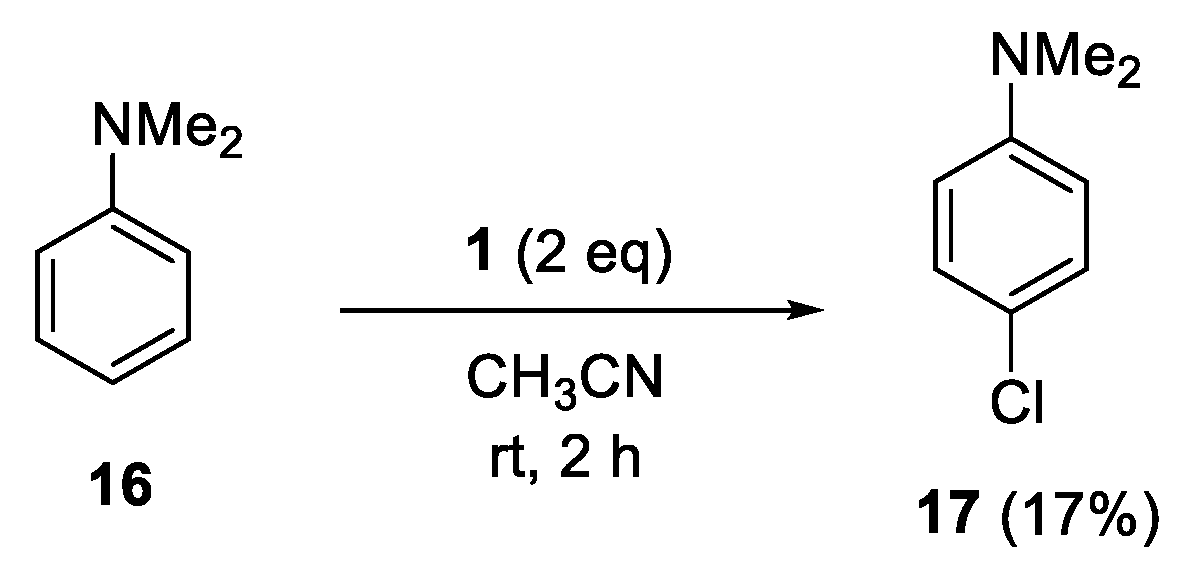
4.10. Reaction of N,N-Dimethylaniline (16)
4.11. X-ray Analysis for 4a
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Veisi, H. Sodium Hypochlorite (NaOCl). Synlett 2007, 2007, 2607–2608. [Google Scholar] [CrossRef]
- Sodium Hypochlorite. Available online: https://www.organic-chemistry.org/chemicals/oxidations/sodiumhypochlorite.shtm (accessed on 13 April 2024).
- Mirafzal, G.A.; Lozeva, A.M. Phase Transfer Catalyzed Oxidation of Alcohols with Sodium Hypochlorite. Tetrahedron Lett. 1998, 39, 7263–7266. [Google Scholar] [CrossRef]
- Fey, T.; Fischer, H.; Bachmann, S.; Albert, K.; Bolm, C. Silica-Supported TEMPO Catalysts: Synthesis and Application in the Anelli Oxidation of Alcohols. J. Org. Chem. 2001, 66, 8154–8159. [Google Scholar] [CrossRef] [PubMed]
- Gonsalvi, L.; Arends, I.W.C.E.; Sheldon, R.A. Selective Ruthenium-Catalyzed Oxidation of 1,2:4,5-Di-O-Isopropylidene-β-D-Fructopyranose and Other Alcohols with NaOCl. Org. Lett. 2002, 4, 1659–1661. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi, Y. 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, A.; Chinnusamy, T.; Cuevas-Yañez, E.; Hilgers, P.; Reiser, O. Combination of Perfluoroalkyl and Triazole Moieties: A New Recovery Strategy for TEMPO. Org. Lett. 2008, 10, 4171–4174. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly Enantioselective Epoxidation Catalysts Derived from l,2-Diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Klawonn, M.; Bhor, S.; Mehltretter, G.; Döbler, C.; Fischer, C.; Beller, M. A Simple and Convenient Method for Epoxidation of Olefins without Metal Catalysts. Adv. Synth. Catal. 2003, 345, 389–392. [Google Scholar] [CrossRef]
- Ooi, T.; Ohara, D.; Tamura, M.; Maruoka, K. Design of New Chiral Phase-Transfer Catalysts with Dual Functions for Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2004, 126, 6844–6845. [Google Scholar] [CrossRef]
- Mehltretter, G.M.; Bhor, S.; Klawonn, M.; Döbler, C.; Sundermeier, U.; Eckert, M.; Militzer, H.C.; Beller, M. A New Practical Method for the Osmium-Catalyzed Dihydroxylation of Olefins Using Bleach as the Terminal Oxidant. Synthesis 2003, 2003, 0295–0301. [Google Scholar] [CrossRef]
- Chellamani, A.; Harikengaram, S. Mechanism of Oxidation of Aryl Methyl Sulfoxides with Sodium Hypochlorite Catalyzed by (Salen)MnIII Complexes. J. Mol. Catal. A Chem. 2006, 247, 260–267. [Google Scholar] [CrossRef]
- Bianchini, J.P.; Cocordano, M. Etude Expérimentale et Théorique de l’addition de l’acide Hypochloreux Sur Les Hydrocarbures Alléniques. Tetrahedron 1970, 26, 3401–3411. [Google Scholar] [CrossRef]
- Moye, C.J.; Sternhell, S. The Degradation of Aromatic Rings: The Action of Hypochlorite on Phenols. Aust. J. Chem. 1966, 19, 2107–2118. [Google Scholar] [CrossRef]
- Strunz, G.M.; Court, A.S.; Komlosay, J. Synthetic Analogues of Cryptosporiopsin: The Action of Hypochlorite on m-Cresol. Tetrahedron Lett. 1969, 10, 3613–3614. [Google Scholar] [CrossRef] [PubMed]
- Favreau, S.; Lizzani-Cuvelier, L.; Loiseau, M.; Dunach, E.; Fellous, R. Novel Synthesis of 3-Oxazolines. Tetrahedron Lett. 2000, 41, 9787–9790. [Google Scholar] [CrossRef]
- Chen, C.Y.; Andreani, T.; Li, H. A Divergent and Selective Synthesis of Isomeric Benzoxazoles from a Single N-Cl Imine. Org. Lett. 2011, 13, 6300–6303. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, C.; Xia, J.B. A Simple Method for the Electrophilic Cyanation of Secondary Amines. Org. Lett. 2014, 16, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.Y.; Chisholm, M.J. Chlorination by Aqueous Sodium Hypochlorite. Can. J. Res. 1946, 24, 208–210. [Google Scholar] [CrossRef]
- Haszeldine, R.N.; Rigby, R.B.; Tipping, A.E. Perfluoroalkyl Derivatives of Sulphur. Part XV. Preparation and Certain Reactions of Methyl Polyfluoroalkyl Sulphoxides and Sulphones and Their Conversion into Polyfluoroalkanesulphonic Acids. J. Chem. Soc. Perkin 1 1973, 676–682. [Google Scholar] [CrossRef]
- Gonsalvi, L.; Arends, I.W.C.E.; Sheldon, R.A. Highly Efficient Use of NaOCl in the Ru-Catalysed Oxidation of Aliphatic Ethers to Esters. Chem. Commun. 2002, 2, 202–203. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, L.; Su, W. Direct Benzylic Oxidation with Sodium Hypochlorite Using a New Efficient Catalytic System: TEMPO/Co(OAc)2. Synlett 2011, 2011, 1435–1438. [Google Scholar] [CrossRef]
- Ruan, L.; Shi, M.; Li, N.; Ding, X.; Yang, F.; Tang, J. Practical Approach for Preparation of Unsymmetric Benzils from β-Ketoaldehydes. Org. Lett. 2014, 16, 733–735. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, B.; Xu, J. Clean and Economic Synthesis of Alkanesulfonyl Chlorides from S-Alkyl Isothiourea Salts via Bleach Oxidative Chlorosulfonation. Synthesis 2014, 46, 225–229. [Google Scholar] [CrossRef]
- Flegeau, E.F.; Harrison, J.M.; Willis, M.C. One-Pot Sulfonamide Synthesis Exploiting the Palladium-Catalyzed Sulfination of Aryl Iodides. Synlett 2016, 27, 101–105. [Google Scholar] [CrossRef]
- Chamorro-Arenas, D.; Osorio-Nieto, U.; Quintero, L.; Herna, L.; Sartillo-Piscil, F. Selective, Catalytic, and Dual C(Sp3)−H Oxidation of Piperazines and Morpholines under Transition-Metal-Free Conditions. J. Org. Chem. 2018, 83, 15333–15346. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tang, Y.; Gao, J.; Rao, G.; Mao, Z. Practical Transition-Metal-Free Protodeboronation of Arylboronic Acids in Aqueous Sodium Hypochlorite. Synlett 2020, 31, 2039–2042. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Sugiyama, Y.; Akiyoshi, M.; Matsunaga, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate Crystals (NaOCl·5H2O): A Convenient and Environmentally Benign Oxidant for Organic Synthesis. Org. Process Res. Dev. 2017, 21, 1925–1937. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Asawa, T.; Sugiyama, Y.; Kimura, Y. Organic Syntheses Using Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) Crystals. J. Synth. Org. Chem. 2020, 78, 11–27. [Google Scholar] [CrossRef]
- Okada, T.; Asawa, T.; Sugiyama, Y.; Kirihara, M.; Iwai, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) Crystals as an Extraordinary Oxidant for Primary and Secondary Alcohols. Synlett 2014, 25, 596–598. [Google Scholar] [CrossRef]
- Okada, T.; Asawa, T.; Sugiyama, Y.; Iwai, T.; Kirihara, M.; Kimura, Y. Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) Crystals; An Effective Re-Oxidant for TEMPO Oxidation. Tetrahedron 2016, 72, 2818–2827. [Google Scholar] [CrossRef]
- Hirashita, T.; Sugihara, Y.; Ishikawa, S.; Naito, Y.; Matsukawa, Y.; Araki, S. Revisiting Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) for the Oxidation of Alcohols in Acetonitrile without Nitroxyl Radicals. Synlett 2018, 29, 2404–2407. [Google Scholar] [CrossRef]
- Okada, T.; Matsumuro, H.; Kitagawa, S.; Iwai, T.; Yamazaki, K.; Kinoshita, Y.; Kimura, Y.; Kirihara, M. Selective Synthesis of Sulfoxides through Oxidation of Sulfides with Sodium Hypochlorite Pentahydrate Crystals. Synlett 2015, 26, 2547–2552. [Google Scholar] [CrossRef]
- Shimazu, H.; Okada, T.; Asawa, T.; Kirihara, M. JP (Japan Kokai Tokkyo Koho) 2017–52730. Chem. Abstr. 2017, 166, 305244. [Google Scholar]
- Kirihara, M.; Osugi, R.; Saito, K.; Adachi, K.; Yamazaki, K.; Matsushima, R.; Kimura, Y. Sodium Hypochlorite Pentahydrate as a Reagent for the Cleavage of Trans-Cyclic Glycols. J. Org. Chem. 2019, 84, 8330–8336. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Mori, H.; Odagiri, T.; Suzuki, K.; Kikkawa, Y.; Osugi, R.; Takizawa, S.; Kimura, Y.; Kirihara, M. A Concise, Catalyst-Free Synthesis of Davis’ Oxaziridines Using Sodium Hypochlorite. SynOpen 2019, 3, 21–25. [Google Scholar] [CrossRef]
- Minakata, S.; Miwa, H.; Yamamoto, K.; Hirayama, A.; Okumura, S. Diastereodivergent Intermolecular 1,2-Diamination of Unactivated Alkenes Enabled by Iodine Catalysis. J. Am. Chem. Soc. 2021, 143, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Miyamoto, K.; Okada, T.; Asawa, T.; Uchiyama, M. Safer Synthesis of (Diacetoxyiodo)Arenes Using Sodium Hypochlorite Pentahydrate. J. Org. Chem. 2018, 83, 14262–14268. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Matsumuro, H.; Iwai, T.; Kitagawa, S.; Yamazaki, K.; Akiyama, T.; Asawa, T.; Sugiyama, Y.; Kimura, Y.; Kirihara, M. An Efficient Method for the Preparation of Sulfonyl Chlorides: Reaction of Disulfides or Thiols with Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) Crystals. Chem. Lett. 2015, 44, 185–187. [Google Scholar] [CrossRef]
- Uyanik, M.; Sasakura, N.; Kuwahata, M.; Ejima, Y.; Ishihara, K. Practical Oxidative Dearomatization of Phenols with Sodium Hypochlorite Pentahydrate. Chem. Lett. 2015, 44, 381–383. [Google Scholar] [CrossRef]
- Kirihara, M.; Odagiri, T.; Asawa, T. JP (Japan Kokai Tokkyo Koho) 2017–52728. Chem. Abstr. 2017, 166, 304950. [Google Scholar]
- Lee, S.M.; Lee, W.G.; Kim, Y.C.; Ko, H. Synthesis and Biological Evaluation of α,β-Unsaturated Lactones as Potent Immunosuppressive Agents. Bioorg. Med. Chem. Lett. 2011, 21, 5726–5729. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.K.; Diaz, M.A.N.; Diaz-Muñoz, G. Lactones: Classification, Synthesis, Biological Activities, and Industrial Applications. Tetrahedron 2021, 84, 132001. [Google Scholar] [CrossRef]
- Hur, J.; Jang, J.; Sim, J.; Hur, J.; Jang, J.; Sim, J.A.; Wawrzé, C. A Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones. Int. J. Mol. Sci. 2021, 22, 2769. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wang, L.L.; Mao, Y.H.; Dong, Y.X.; Liu, B.; Zhu, G.F.; Yang, Y.Y.; Guo, B.; Tang, L.; Zhang, J.Q. Iron-Catalyzed Radical Annulation of Unsaturated Carboxylic Acids with Disulfides for the Synthesis of γ-Lactones. J. Org. Chem. 2021, 86, 8620–8629. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Rehbein, J.; Wirth, T. Facile Oxidative Rearrangements Using Hypervalent Iodine Reagents. ChemistryOpen 2012, 1, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Ha, J.; Woo, J.; Kim, D.Y. Electrochemical Oxidative Bromolactonization of Unsaturated Carboxylic Acids with Sodium Bromide: Synthesis of Bromomethylated γ-Lactones. Tetrahedron Lett. 2022, 88, 153567. [Google Scholar] [CrossRef]
- Campbell, M.L.; Rackley, S.A.; Giambalvo, L.N.; Whitehead, D.C. Vanadium (V) Oxide Mediated Bromolactonization of Alkenoic Acids. Tetrahedron 2015, 71, 3895–3902. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L.; Santi, C. Selenium Catalysed Conversion of β,γ-Unsaturated Acids into Butenolides. Synlett 1993, 1993, 798–800. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Taka, H.; Oishi, A.; Ikeda, Y.; Taguchi, Y.; Fujie, K.; Saeki, T.; Sakuma, M. Efficient Catalytic Selenolactonization-Deselenenylation Reactions Using Various Organoselenium Catalysts and Their Application to Solid-Phase Conversion. Synlett 2000, 2000, 1509–1511. [Google Scholar] [CrossRef]
- Fujita, K.I.; Hashimoto, S.; Oishi, A.; Taguchi, Y. Intramolecular Oxyselenenylation and Deselenenylation Reactions in Water, Conducted by Employing Polymer-Supported Arylselenenyl Bromide. Tetrahedron Lett. 2003, 44, 3793–3795. [Google Scholar] [CrossRef]
- Richardson, R.D.; Zayed, J.M.; Altermann, S.; Smith, D.; Wirth, T. Tetrafluoro-IBA and-IBX: Hypervalent Iodine Reagents. Angew. Chem. Int. Ed. 2007, 46, 6529–6532. [Google Scholar] [CrossRef]
- Shah, A.U.H.A.; Khan, Z.A.; Choudhary, N.; Lohölter, C.; Schäfer, S.; Marie, G.P.L.; Farooq, U.; Witulski, B.; Wirth, T. Iodoxolone-Based Hypervalent Iodine Reagents. Org. Lett. 2009, 11, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Jun He, R.; Chun Zhu, B.; Wang, Y.G. Lewis Base-Catalyzed Electrophilic Lactonization of Selenyl Bromide Resin and Facile Solid-Phase Synthesis of Furan-2(5H)-One Derivatives. Appl. Organomet. Chem. 2014, 28, 523–528. [Google Scholar] [CrossRef]
- Kawamata, Y.; Hashimoto, T.; Maruoka, K. A Chiral Electrophilic Selenium Catalyst for Highly Enantioselective Oxidative Cyclization. J. Am. Chem. Soc. 2016, 138, 5206–5209. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Li, Y.; Liu, Y.; Sun, B.; Yang, S.; Tian, H. Synthesis of Butenolides by Reactions of 3-Alkenoic Acids with Diphenyl Sulfoxide/Oxalyl Chloride. Flavour. Fragr. J. 2018, 33, 397–404. [Google Scholar] [CrossRef]
- Ding, R.; Liu, Y.; Liu, L.; Li, H.; Tao, S.; Sun, B.; Tian, H. A Facile Synthesis of γ-Butenolides via Cyclization of 3-Alkenoic Acids with Dimethyl Sulfoxide and Oxalyl Bromide. Synth. Commun. 2019, 49, 3001–3007. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and Anticancer Activity of Novel Halolactones with β-Aryl Substituents from Simple Aromatic Aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Tan, C.K.; Er, J.C.; Yeung, Y.Y. Synthesis of Chiral Butenolides Using Amino-Thiocarbamate-Catalyzed Asymmetric Bromolactonization. Tetrahedron Lett. 2014, 55, 1243–1246. [Google Scholar] [CrossRef]
- Garnier, J.M.; Robin, S.; Rousseau, G. An Approach to Enantioselective 5-Endo Halo-Lactonization Reactions. Eur. J. Org. Chem. 2007, 2007, 3281–3291. [Google Scholar] [CrossRef]
- Ding, R.; Lan, L.; Li, S.; Liu, Y.; Yang, S.; Tian, H.; Sun, B. A Novel Method for the Chlorolactonization of Alkenoic Acids Using Diphenyl Sulfoxide/Oxalyl Chloride. Synthesis 2018, 50, 2555–2566. [Google Scholar] [CrossRef]
- Ángel, A.Y.B.; Bragança, E.F.; Alberto, E.E. Dichlorination of Alkenes Using 1,3-Dichloro-5,5-Dimethylhydantoin and ZnCl2. ChemistrySelect 2019, 4, 11548–11552. [Google Scholar] [CrossRef]
- Badetti, E.; Romano, F.; Marchiò, L.; Taşkesenlioǧlu, S.; Daştan, A.; Zonta, C.; Licini, G. Effective Bromo and Chloro Peroxidation Catalysed by Tungsten(VI) Amino Triphenolate Complexes. Dalton Trans. 2016, 45, 14603–14608. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.J.; Milcent, T.; Crousse, B. Regioselective Halogenation of Arenes and Heterocycles in Hexafluoroisopropanol. J. Org. Chem. 2018, 83, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Saccone, M.; Pfletscher, M.; Kather, S.; Wölper, C.; Daniliuc, C.; Mezger, M.; Giese, M. Improving the Mesomorphic Behaviour of Supramolecular Liquid Crystals by Resonance-Assisted Hydrogen Bonding. J. Mater. Chem. C Mater. 2019, 7, 8643–8648. [Google Scholar] [CrossRef]
- Xu, H.; Hu, L.; Zhu, G.; Zhu, Y.; Wang, Y.; Wu, Z.G.; Zi, Y.; Huang, W. DABCO as a Practical Catalyst for Aromatic Halogenation with N-Halosuccinimides. RSC Adv. 2022, 12, 7115–7119. [Google Scholar] [CrossRef] [PubMed]
- Markushyna, Y.; Teutloff, C.; Kurpil, B.; Cruz, D.; Lauermann, I.; Zhao, Y.; Antonietti, M.; Savateev, A. Halogenation of Aromatic Hydrocarbons by Halide Anion Oxidation with Poly(Heptazine Imide) Photocatalyst. Appl. Catal. B 2019, 248, 211–217. [Google Scholar] [CrossRef]
- Franco, J.U.; Ell, J.R.; Hilton, A.K.; Hammons, J.C.; Olmstead, M.M. C60Cl6, C60Br8 and C60(NO2)6 as Selective Tools in Organic Synthesis. Fuller. Nanotub. Carbon Nanostructures 2009, 17, 349–360. [Google Scholar] [CrossRef]
- Kamigata, N.; Satoh, T.; Yoshida, M.; Matsuyama, H.; Kameyama, M. Regioselective Para Halogenation of Substituted Benzenes with Benzeneseleninyl Chloride and Aluminum Halide. Bull. Chem. Soc. Jpn. 1988, 61, 2226–2228. [Google Scholar] [CrossRef]
- Sivey, J.D.; Roberts, A.L. Assessing the Reactivity of Free Chlorine Constituents Cl2, Cl2O, and HOCl Toward Aromatic Ethers. Environ. Sci. Technol. 2012, 46, 2141–2147. [Google Scholar] [CrossRef]
- Brown, D.M.; Niyomura, O.; Wirth, T. Catalytic Addition-Elimination Reactions Towards Butenolides. Phosphorus Sulfur Silicon 2008, 183, 1026–1035. [Google Scholar] [CrossRef]
- Alexander, T.S.; Clay, T.J.; Maldonado, B.; Nguyen, J.M.; Martin, D.B.C. Comparative Studies of Palladium and Copper-Catalysed g-Arylation of Silyloxy Furans with Diaryliodonium Salts. Tetrahedron 2019, 75, 2229–2238. [Google Scholar] [CrossRef]
- Clawson, P.; Lunn, P.M.; Whiting, D.A. Synthetic Studies on O-Heterocycles via Cycloadditions. Part 1. Photochemical (Electron Transfer Sensitised) C-C Cleavage of Diaryloxiranes. J. Chem. Soc. Perkin 1 1990, 153–157. [Google Scholar] [CrossRef]
- Genovese, S.; Epifano, F.; Pelucchini, C.; Procopio, A.; Curini, M. Ytterbium Triflate Catalyzed Synthesis of Chlorinated Lactones. Tetrahedron Lett. 2010, 51, 5992–5995. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, M.; Xu, Y.; Yan, J. A Novel One-Pot Sulfonyloxylactonization of Alkenoic Acids Mediated by Hypervalent Iodine Species Generated in Situ from Ammonium Iodide. Helv. Chim. Acta 2014, 97, 137–145. [Google Scholar] [CrossRef]
- Pimenta, L.S.; Gusevskaya, E.V.; Alberto, E.E. Intermolecular Halogenation/Esterification of Alkenes with N-Halosuccinimide and Acetic Acid Catalyzed by 1,4-Diazabicyclo[2.2.2]Octane. Adv. Synth. Catal. 2017, 359, 2297–2303. [Google Scholar] [CrossRef]
- Deng, Y.; Wei, X.J.; Wang, X.; Sun, Y.; Noël, T. Iron-Catalyzed Cross-Coupling of Alkynyl and Styrenyl Chlorides with Alkyl Grignard Reagents in Batch and Flow. Chem. Eur. J. 2019, 25, 14532–14535. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, Z.; Stahl, S.S. Regioselective Copper-Catalyzed Chlorination and Bromination of Arenes with O2 as the Oxidant. Chem. Commun. 2009, 42, 6460–6462. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Ma, Z.; Gao, X.; Liu, Z. B(C6F5)3-Catalyzed Methylation of Amines Using CO2 as a C1 Building Block. Green Chem. 2015, 17, 4189–4193. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Foundations and Advances SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]

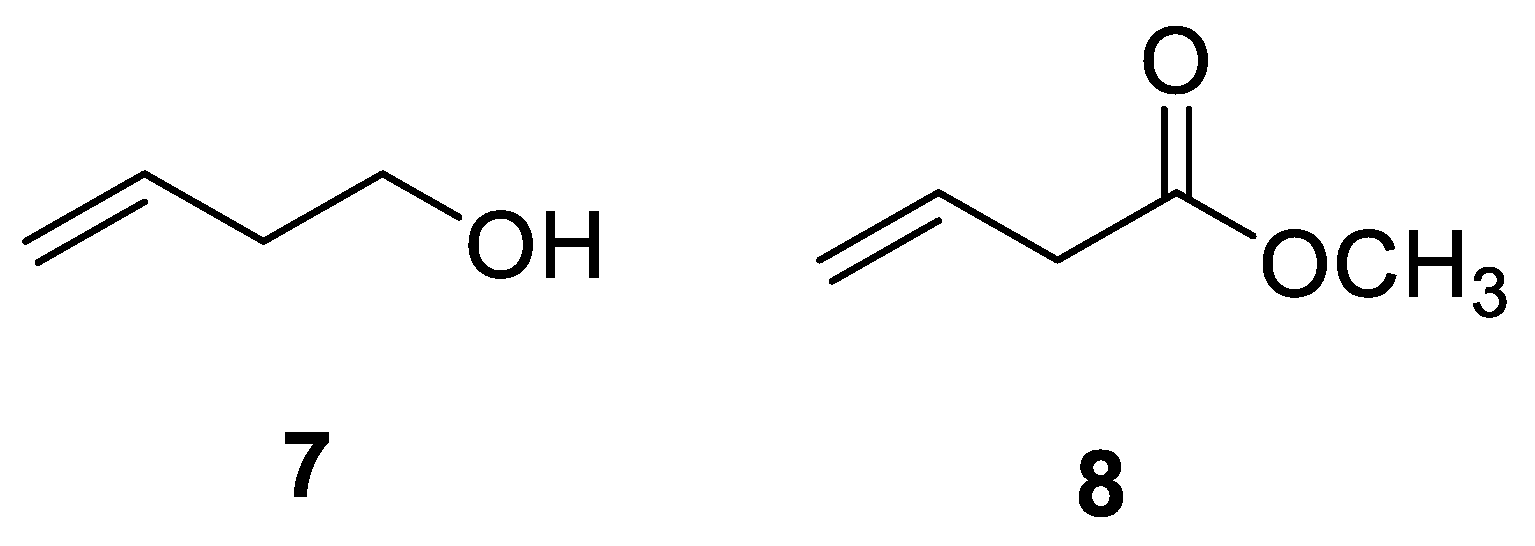


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
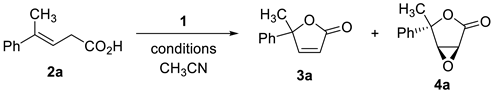 | |||||
|---|---|---|---|---|---|
| Entry | NaClO·5H2O (1) | Temp | Reaction Time (h) | Yields of 3a (%) b | Yields of 4a (%) b |
| 1 | 1 eq | 0 °C | 1 | trace | – |
| 2 | 2 eq | 0 °C | 1 | 53 | – |
| 3 | 2 eq | 0 °C | 3 | 36 | – |
| 4 | 3 eq | 0 °C | 3 | 45 | 7 |
| 5 | 5 eq | 0 °C | 3 | 17 | 5 |
| 6 | 1 eq | rt | 1 | 48 | – |
| 7 | 2 eq | rt | 1 | 62 | – |
| 8 | 3 eq | rt | 1 | 37 | 25 |
| 9 | 5 eq | rt | 24 | 26 | 20 |
 | |||
|---|---|---|---|
| Entry | Solvents | Yields of 3a (%) b | Yields of 5a (%) b |
| 1 | CH3CN | 62 | – |
| 2 | MeOH | 61 | – |
| 3 | DMF | 59 | – |
| 4 | EtOH | 39 | – |
| 5 | Acetone | 25 | – |
| 6 | Hexane | 18 | – |
| 7 | Toluene | 17 | – |
| 8 | DCM | 11 | – |
| 9 | H2O | 24 | trace c |
| 10 | Buffer pH 6.9 d | 21 | 10 |
| 11 | Buffer pH 4.0 e | 48 | 25 |
| 12 | Buffer pH 4.0—Et2O | 17 | 32 |
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | R | Conditions | Yield of 3 b | Side Products (Yields) b |
| 1 | 2b | Ph | rt, 1 h | 21% | 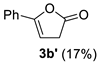 |
| rt, 6 h | – | (30%) | |||
| rt, 1 h c | 23% | (trace) | |||
| 2 | 2c | 4-CH3O-C6H4 | 0 °C, 3 h | 56% | |
| 3 | 2d | H | 0 °C, 1 h d | 0% | 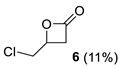 |
| 4 | 2e | CH3CH2 | rt, 1 d d,e | 16% |  |
| 5 | 2f | PhCH2 | 0 °C, 2 h d | 8% | 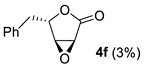 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwaoka, M.; Shimada, R.; Kuroda, M.; Ikeda, T.; Alberto, E.E. Sodium Hypochlorite Pentahydrate as a Chlorinating Reagent: Application to the Tandem Conversion of β,γ-Unsaturated Carboxylic Acids to α,β-Unsaturated Lactones. Processes 2024, 12, 1102. https://doi.org/10.3390/pr12061102
Iwaoka M, Shimada R, Kuroda M, Ikeda T, Alberto EE. Sodium Hypochlorite Pentahydrate as a Chlorinating Reagent: Application to the Tandem Conversion of β,γ-Unsaturated Carboxylic Acids to α,β-Unsaturated Lactones. Processes. 2024; 12(6):1102. https://doi.org/10.3390/pr12061102
Chicago/Turabian StyleIwaoka, Michio, Reo Shimada, Masaki Kuroda, Takehito Ikeda, and Eduardo E. Alberto. 2024. "Sodium Hypochlorite Pentahydrate as a Chlorinating Reagent: Application to the Tandem Conversion of β,γ-Unsaturated Carboxylic Acids to α,β-Unsaturated Lactones" Processes 12, no. 6: 1102. https://doi.org/10.3390/pr12061102