
Inverse Method-Based Kinetic Modelling and Process Optimization of Reverse-Phase Chromatography for Molnupiravir Synthesis


Abstract
:1. Introduction
2. Materials and Methods
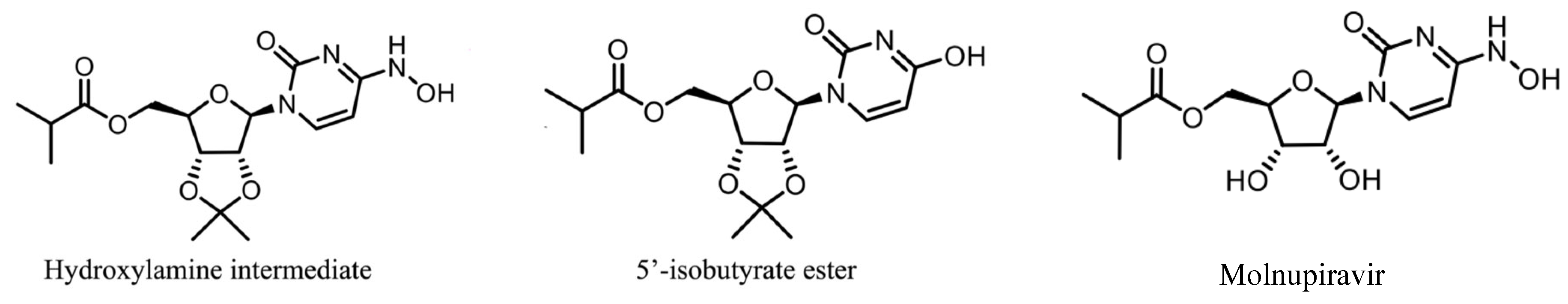
2.1. Process Description
2.2. Experimental Setup
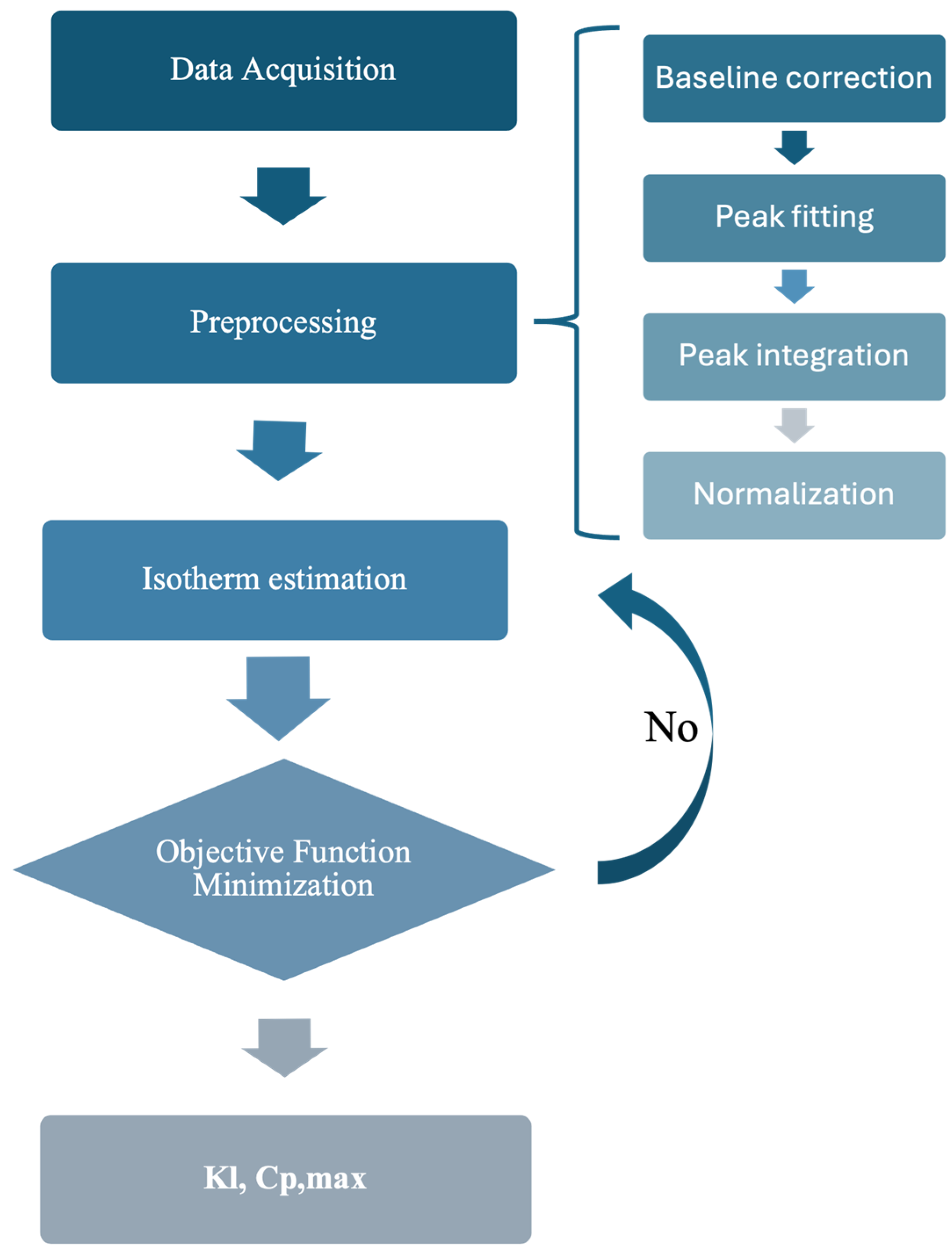
2.3. Adsorption Isotherm Determination
2.4. Chromatographic Model Simulation
3. Results and Discussions
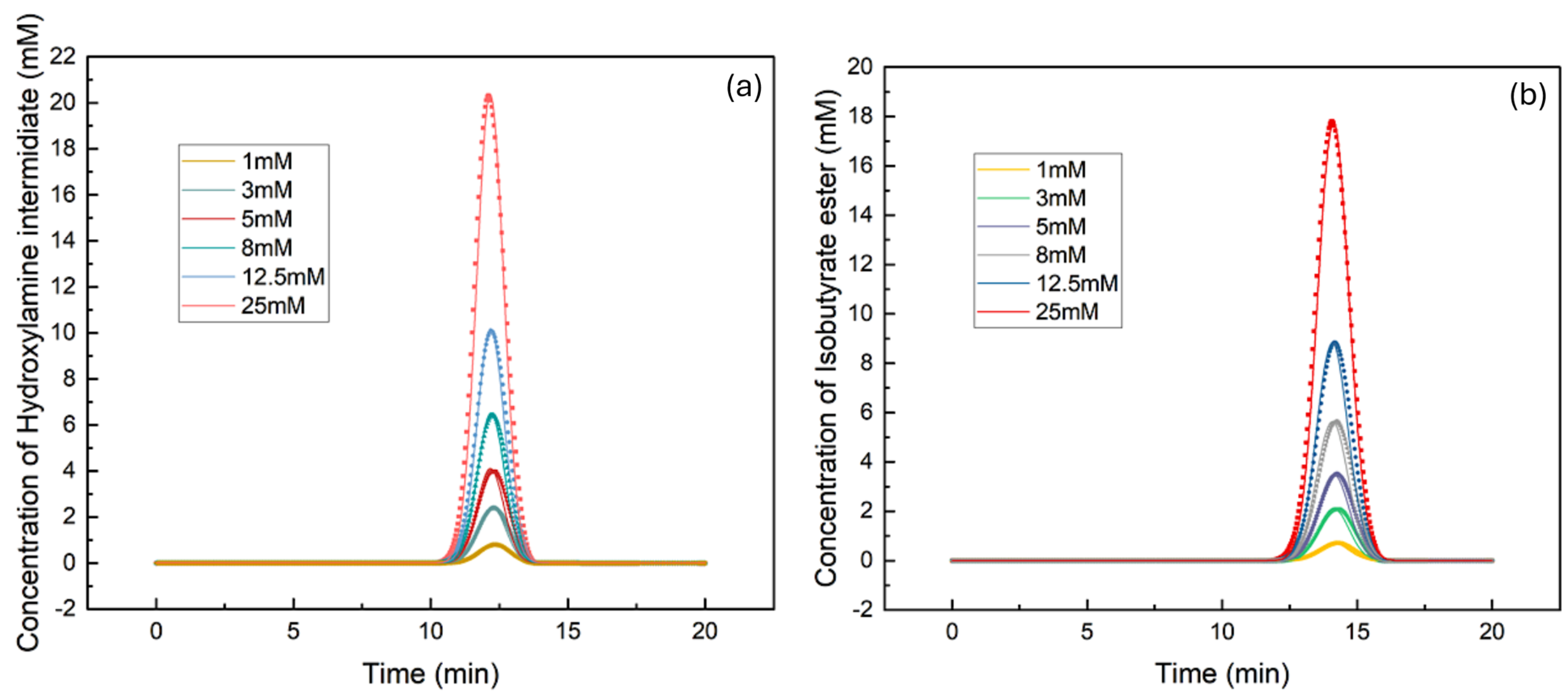
3.1. Determination of Isotherm Constants for the Single–Component Data Using the Inverse Method
3.2. Effects of Dispersivity Coefficient on Chromatography Profile
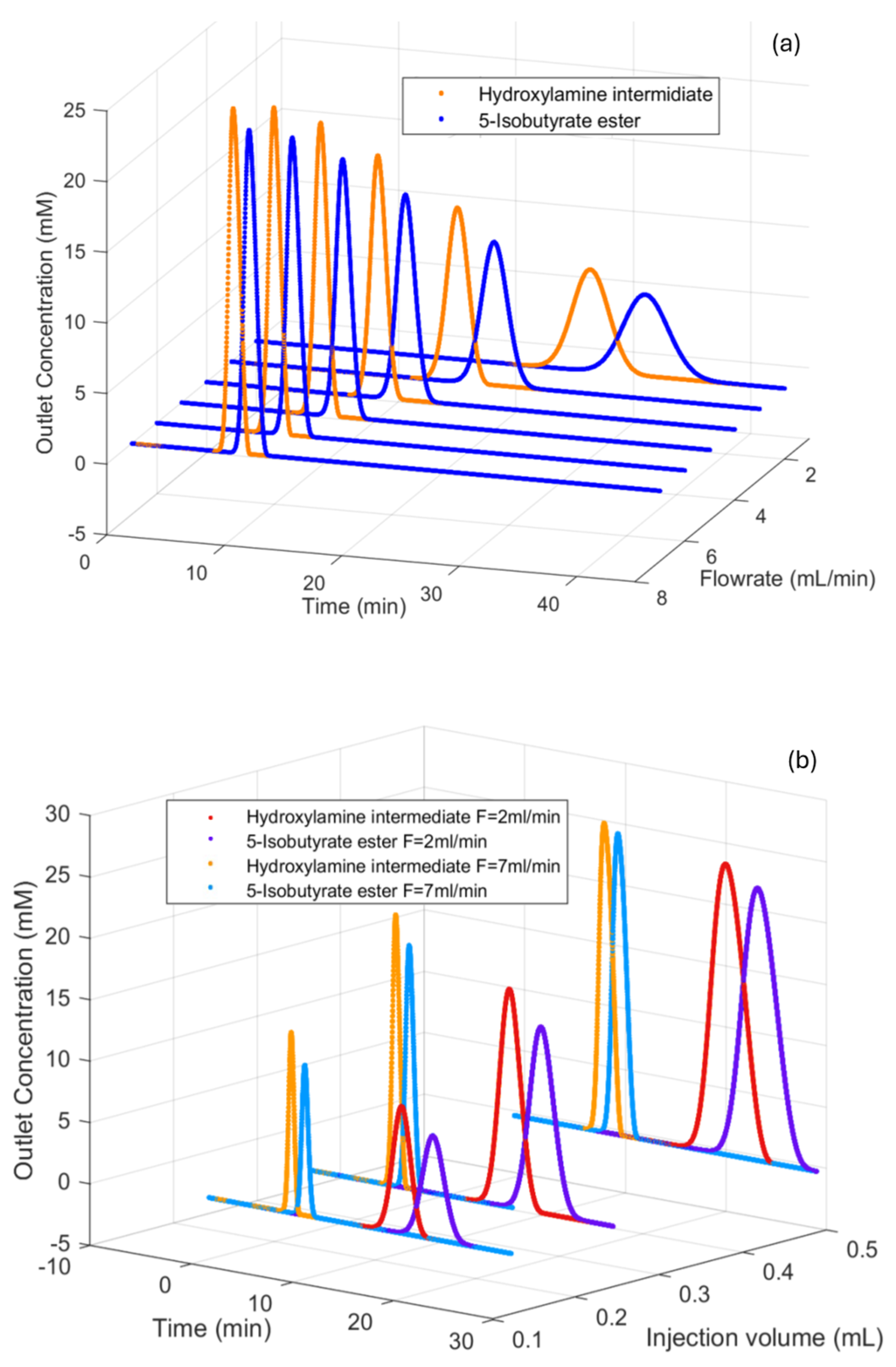
3.3. Effects of Injection Volume on Chromatography Profile
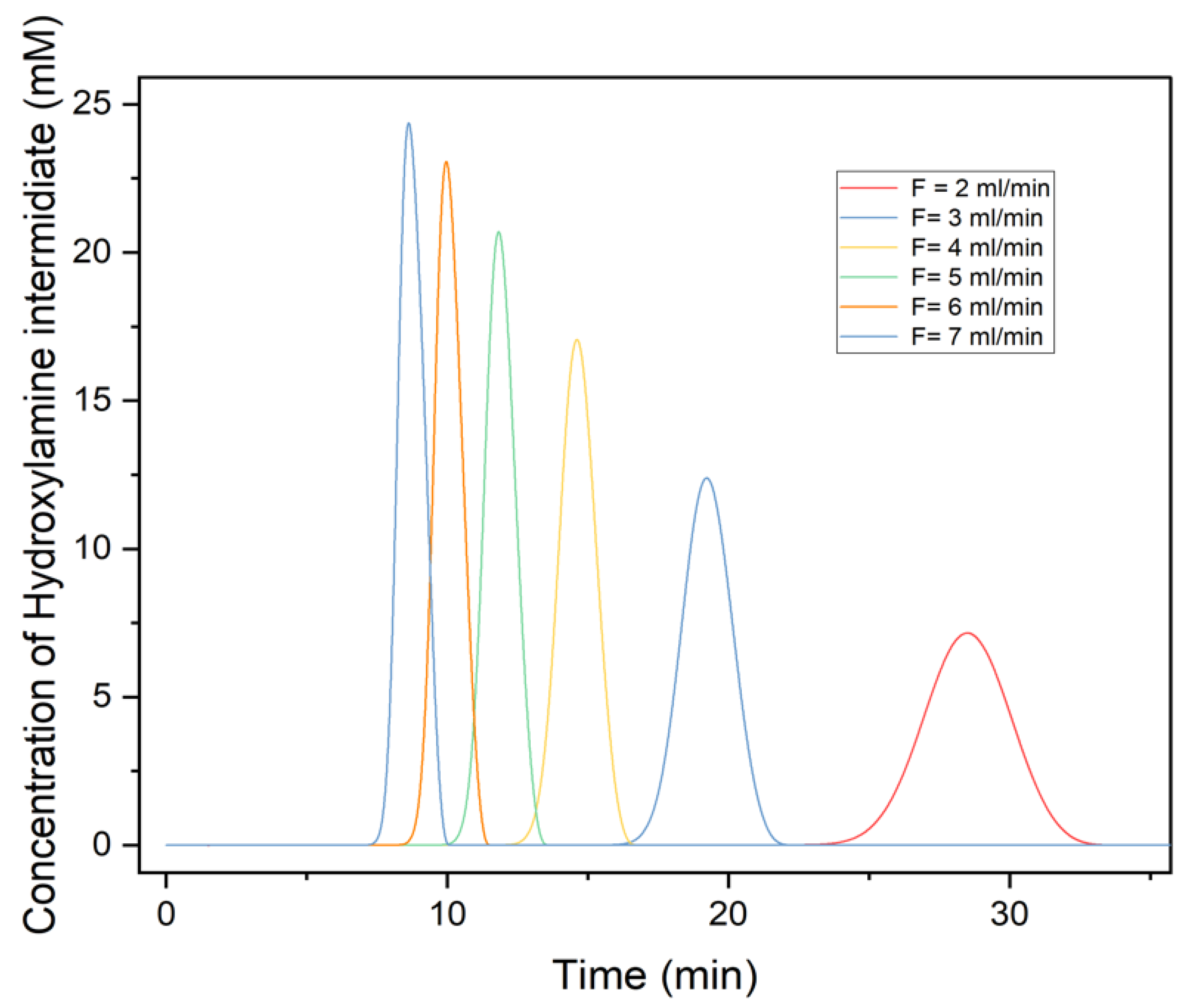
3.4. Effects of Flowrate on Chromatography Profile
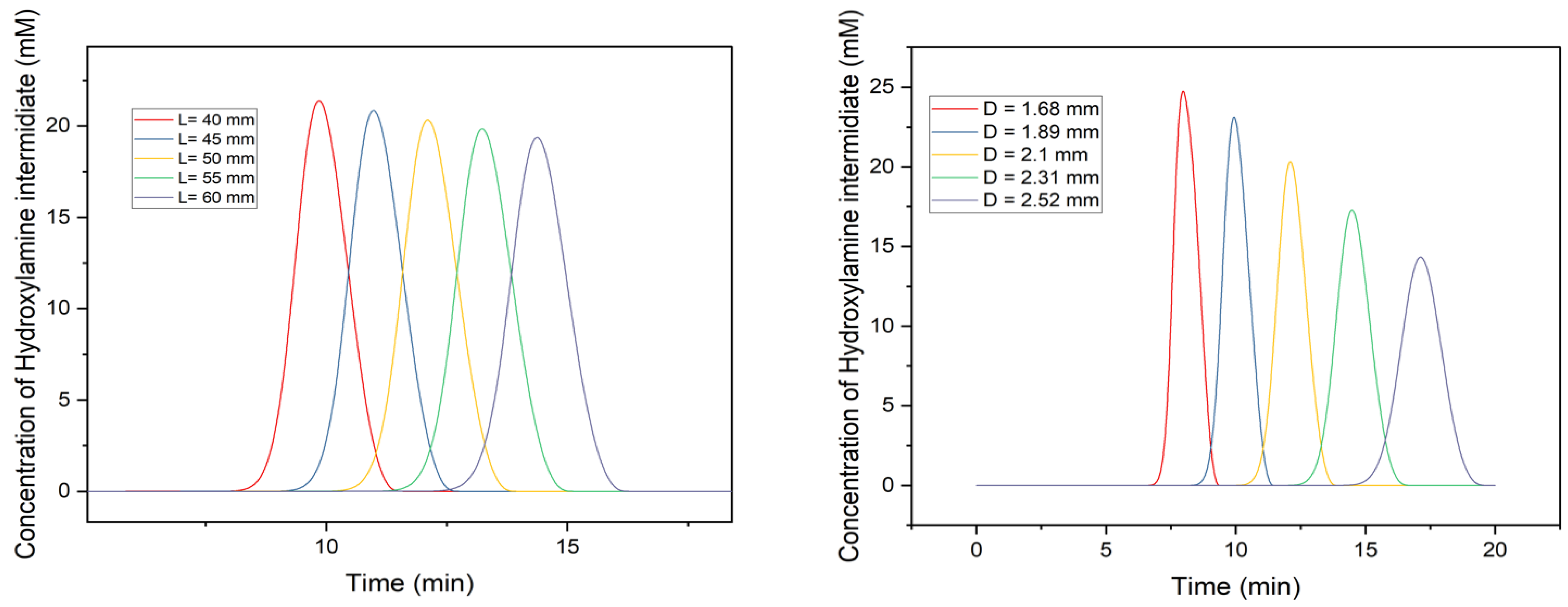
3.5. Effects of Column’s Geometry on Chromatography Profile
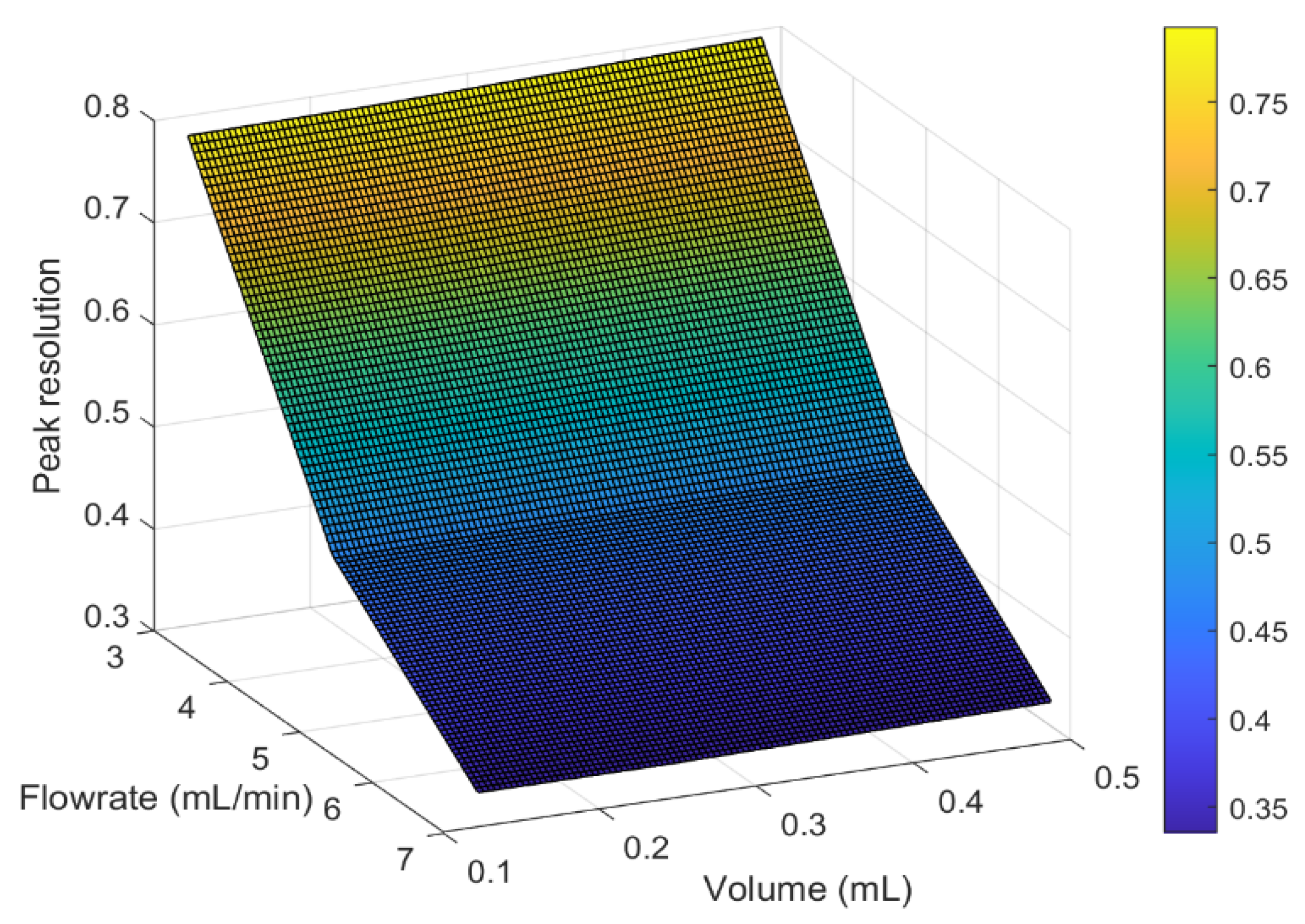
3.6. Investigating the Effect of Operational Conditions on Peak Resolution
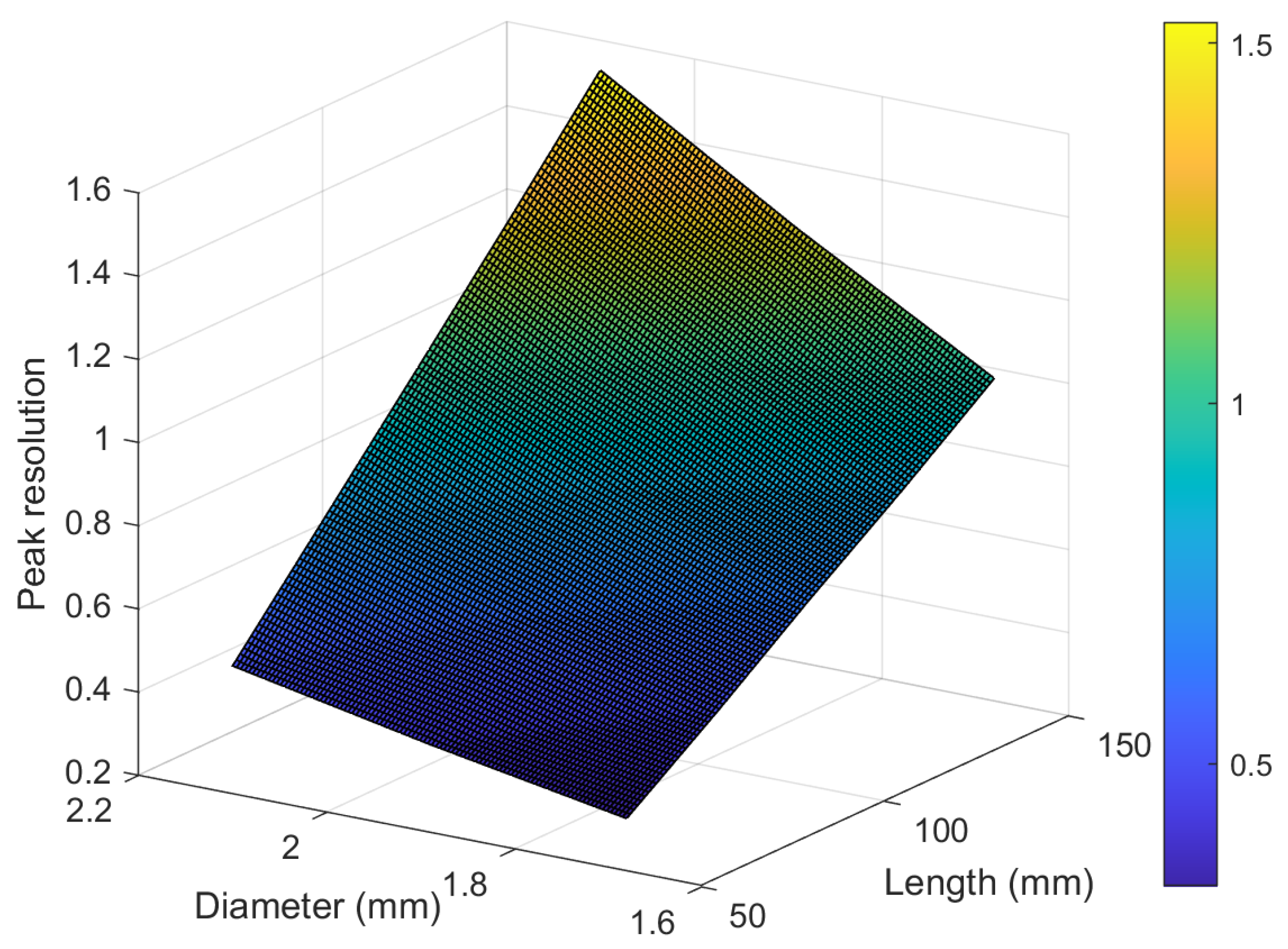
3.7. Investigating the Effect of Geometry on Peak Resolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| Symbol | Variable | Units |
| Cp,max | Maximum adsorption capacity of species i on the adsorbent | mol/m3 |
| ci | Concentration of species i in the liquid | mol/L |
| D | Effective dispersion coefficient | m2/s |
| Df,i | Diffusion coefficient of species i in the liquid phase | m2/s |
| εp | Porosity of the porous medium | - |
| Ji | Total mass transfer flux of species i | mol/(s·m2) |
| kB | Boltzmann constant | J/K |
| KL,i | Langmuir adsorption equilibrium constant for species i | L/mol |
| L | Column length | m |
| N | Number of theoretical plates | - |
| n | Outward normal vector at the column wall | - |
| ri | Residual between simulation and measurement for species i | - |
| ρ | Density of the solid particles | kg/m3 |
| R | Column radius | m |
| r | Hydrodynamic radius of particle | m |
| tR | Peak retention time | s |
| T | Temperature | K |
| t | Time | s |
| u | Velocity of the liquid | m/s |
| w | Base peak width | s |
| z | Axial coordinate along the column length | m |
| η | Solvent viscosity | Pa·s |
| Abbreviations | Variable | |
| API | Active pharmaceutical ingredient | |
| C18 | A type of bonded phase employed in the column | |
| C4 | Butyl silica | |
| C8 | Octyl silica | |
| CM | Continuous manufacturing | |
| ECP | Elution by characteristic point | |
| ED | Equilibrium-dispersive model | |
| EUA | Emergency Use Authorization | |
| FA | Frontal analysis | |
| GR | General Rate model | |
| MS | Mass spectrometry | |
| OBD | Optimized for Broad Design | |
| PM | Perturbation | |
| POR | Lumped pore diffusion model | |
| RPC | Reversephase chromatography | |
| RPLC | Reversed-phase liquid chromatography | |
| TD | Transport-dispersive model | |
| UHPLC | Ultra-high-performance liquid chromatography | |
| UV | Ultraviolet |
References
- Saxena, S.K.; Kumar, S.; Ansari, S.; Paweska, J.T.; Maurya, V.K.; Tripathi, A.K.; Abdel-Moneim, A.S. Transmission dynamics and mutational prevalence of the novel Severe acute respiratory syndrome coronavirus-2 Omicron Variant of Concern. J. Med. Virol. 2022, 94, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Al Meslamani, A.Z. Advancing towards a worldwide healthcare system in the post-COVID-19 era: Benefits and barriers to international collaboration in healthcare. J. Med. Econ. 2023, 26, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A., 2nd; Eron, J.J., Jr.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Xie, Y.; Zhu, F.; Gong, X.; Liu, Y.; Xue, H.; Aisa, H.A.; Shen, J. “One-Pot” Synthesis of Molnupiravir from Cytidine. Org. Process Res. Dev. 2022, 26, 358–364. [Google Scholar] [CrossRef]
- Zarenezhad, E.; Marzi, M. Review on molnupiravir as a promising oral drug for the treatment of COVID-19. Med. Chem. Res. 2022, 31, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Dey, R.; Nayak, S.; Das, P.; Yadav, S. Short Synthesis of Molnupiravir (EIDD-2801) via a Thionated Uridine Intermediate. ACS Omega 2021, 6, 28366–28372. [Google Scholar] [CrossRef] [PubMed]
- Cruz, G.; Acosta, J.; Del Arco, J.; Clemente-Suarez, V.J.; Deroncele, V.; Fernández-Lucas, J. Enzyme-mediated synthesis of Molnupiravir: Paving the way for the application of biocatalysis in pharmaceutical industry. ChemCatChem 2022, 14, e202200140. [Google Scholar] [CrossRef]
- Paymode, D.J.; Vasudevan, N.; Ahmad, S.; Kadam, A.L.; Cardoso, F.S.P.; Burns, J.M.; Cook, D.W.; Stringham, R.W.; Snead, D.R. Toward a Practical, Two-Step Process for Molnupiravir: Direct Hydroxamination of Cytidine Followed by Selective Esterification. Org. Process Res. Dev. 2021, 25, 1822–1830. [Google Scholar] [CrossRef]
- Steiner, A.; Znidar, D.; Otvos, S.B.; Snead, D.R.; Dallinger, D.; Kappe, C.O. A High-Yielding Synthesis of EIDD-2801 from Uridine. Eur. J. Org. Chem. 2020, 2020, 6736–6739. [Google Scholar] [CrossRef]
- Vasudevan, N.; Ahlqvist, G.P.; McGeough, C.P.; Paymode, D.J.; Cardoso, F.S.P.; Lucas, T.; Dietz, J.P.; Opatz, T.; Jamison, T.F.; Gupton, F.B.; et al. A concise route to MK-4482 (EIDD-2801) from cytidine. Chem. Commun. 2020, 56, 13363–13364. [Google Scholar] [CrossRef]
- Dyr, J.E.; Suttnar, J. Separation used for purification of recombinant proteins. J. Chromatogr. B Biomed. Sci. Appl. 1997, 699, 383–401. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Boysen, R.I.; Yang, Y.; Hearn, M.T. Aqueous normal-phase chromatography using silica-hydride-based stationary phases. TrAC Trends Anal. Chem. 2013, 42, 64–73. [Google Scholar] [CrossRef]
- Rogers, L.A.; Crews, K.E.; Long, S.G.; Patterson, K.M.; McCune, J.E. Evaluation of chromatographic methods for drug products containing polar and non-polar molecules using reversed phase, hydrophilic interaction, and ion exchange chromatography. J. Liq. Chromatogr. Relat. Technol. 2009, 32, 2246–2264. [Google Scholar] [CrossRef]
- Kirkland, J. Development of some stationary phases for reversed-phase HPLC. J. Chromatogr. A 2004, 1060, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P.; Janás, P.; Škeříková, V.; Urban, J. Effect of water on the retention on diol and amide columns in hydrophilic interaction liquid chromatography. J. Sep. Sci. 2017, 40, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Fausnaugh, J.; Kennedy, L.; Regnier, F. Comparison of hydrophobic-interaction and reversed-phase chromatography of proteins. J. Chromatogr. A 1984, 317, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Vailaya, A. Fundamentals of reversed phase chromatography: Thermodynamic and exothermodynamic treatment. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 965–1054. [Google Scholar] [CrossRef]
- Andrzejewska, A.; Kaczmarski, K.; Guiochon, G. Theoretical study of the accuracy of the pulse method, frontal analysis, and frontal analysis by characteristic points for the determination of single component adsorption isotherms. J. Chromatogr. A 2009, 1216, 1067–1083. [Google Scholar] [CrossRef]
- Lisec, O.; Hugo, P.; Seidel-Morgenstern, A. Frontal analysis method to determine competitive adsorption isotherms. J. Chromatogr. A 2001, 908, 19–34. [Google Scholar] [CrossRef]
- Jacobson, J.; Frenz, J.; Horváth, C. Measurement of adsorption isotherms by liquid chromatography. J. Chromatogr. A 1984, 316, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.-L. Design, Modeling, and Optimization of a Multicomponent Separation Process for Insulin Purification Using Reversed Phase Chromatography. Ph.D. Thesis, Purdue University, West Lafayette, IN, USA, 2008. [Google Scholar]
- Samuelsson, J.; Undin, T.; Fornstedt, T. Expanding the elution by characteristic point method for determination of various types of adsorption isotherms. J. Chromatogr. A 2011, 1218, 3737–3742. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Stanley, B.J.; Guiochon, G. Theoretical study of the accuracy and precision of the measurement of single-component isotherms by the elution by characteristic point method. J. Chromatogr. A 1994, 659, 27–41. [Google Scholar] [CrossRef]
- De Luca, C.; Felletti, S.; Macis, M.; Cabri, W.; Lievore, G.; Chenet, T.; Pasti, L.; Morbidelli, M.; Cavazzini, A.; Catani, M. Modeling the nonlinear behavior of a bioactive peptide in reversed-phase gradient elution chromatography. J. Chromatogr. A 2020, 1616, 460789. [Google Scholar] [CrossRef]
- Felinger, A.; Cavazzini, A.; Guiochon, G. Numerical determination of the competitive isotherm of enantiomers. J. Chromatogr. A 2003, 986, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, L.; Xu, G.; Yu, W.; Ray, A.K. Determination of competitive adsorption isotherm of enantiomers on preparative chromatographic columns using inverse method. J. Chromatogr. A 2013, 1273, 49–56. [Google Scholar] [CrossRef]
- Arnell, R.; Forssén, P.; Fornstedt, T. Accurate and rapid estimation of adsorption isotherms in liquid chromatography using the inverse method on plateaus. J. Chromatogr. A 2005, 1099, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F. Perspective on the future approaches to predict retention in liquid chromatography. Anal. Chem. 2021, 93, 5653–5664. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yu, B.; Zhang, Y.; Gao, X.; Zhu, L.; Ma, T.; Yang, H. Bioactivity-Guided Fractionation of an Antidiarrheal Chinese Herb Rhodiola kirilowii (Regel) Maxim Reveals (-)–Epicatechin-3-Gallate and (-)–Epigallocatechin-3-Gallate as Inhibitors of Cystic Fibrosis Transmembrane Conductance Regulator. PLoS ONE 2015, 10, e0119122. [Google Scholar] [CrossRef]
- Arafah, R.S.; Ribeiro, A.E.; Rodrigues, A.E.; Pais, L.S. Separation of nadolol racemates by high pH reversed-phase preparative chromatography. Sep. Purif. Technol. 2020, 233, 116018. [Google Scholar] [CrossRef]
- Dutta, B.K. Principles of Mass Transfer and Seperation Processes; PHI Learning Pvt. Ltd.: Delhi, India, 2007. [Google Scholar]
- Islam, M.A.; Chowdhury, M.A.; Mozumder, M.S.I.; Uddin, M.T. Langmuir adsorption kinetics in liquid media: Interface reaction model. ACS Omega 2021, 6, 14481–14492. [Google Scholar] [CrossRef] [PubMed]
- Boonen, J.; D’Hondt, M.; Veryser, L.; Peremans, K.; Burvenich, C.; Spiegeleer, B.D. A critical quality parameter in quantitative fused-core chromatography: The injection volume. J. Pharm. Anal. 2013, 3, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Yang, Z.; Liang, Y.; Fan, W.; Ding, Q. Effects of injection volume on chromatographic features and resolution in the process of counter-current chromatography. J. Chromatogr. A 2013, 1277, 7–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physics | Inlet (z = 0) | Outlet (L = 50 mm) | Adsorber Wall (R = 0.0465 in) | Initial Condition |
|---|---|---|---|---|
| Mass transfer | c(t, z = 0) = u(t) | No flux | ci = 0 |
| On Component | KL (m3/mol) | Cp,max (mol/kg) | FSSR |
|---|---|---|---|
| Hydroxylamine intermediate | 0.00063409 | 645.24 | 0.9894 |
| 5-Isobutyrate ester | 0.00063756 | 752.25 | 0.9941 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kritikos, A.; Singh, R.; Muzzio, F.; Tsilomelekis, G. Inverse Method-Based Kinetic Modelling and Process Optimization of Reverse-Phase Chromatography for Molnupiravir Synthesis. Processes 2024, 12, 1273. https://doi.org/10.3390/pr12061273
Kritikos A, Singh R, Muzzio F, Tsilomelekis G. Inverse Method-Based Kinetic Modelling and Process Optimization of Reverse-Phase Chromatography for Molnupiravir Synthesis. Processes. 2024; 12(6):1273. https://doi.org/10.3390/pr12061273
Chicago/Turabian StyleKritikos, Athanasios, Ravendra Singh, Fernando Muzzio, and George Tsilomelekis. 2024. "Inverse Method-Based Kinetic Modelling and Process Optimization of Reverse-Phase Chromatography for Molnupiravir Synthesis" Processes 12, no. 6: 1273. https://doi.org/10.3390/pr12061273