
Computational Insight of Oleracone L, Portulacatone B, and Portulacatal from Portulaca oleracea L. as Potential Anticholinesterase Inhibitors for Alzheimer’s

Abstract
:1. Introduction
2. Methodology
2.1. ADME and Physicochemical Predictions
2.2. Molecular Docking Simulation
2.3. Molecular Dynamic Simulation
2.4. MM-PBSA Calculation
3. Results
3.1. ADME and Physicochemical Predictions
3.2. Molecular Docking Simulation
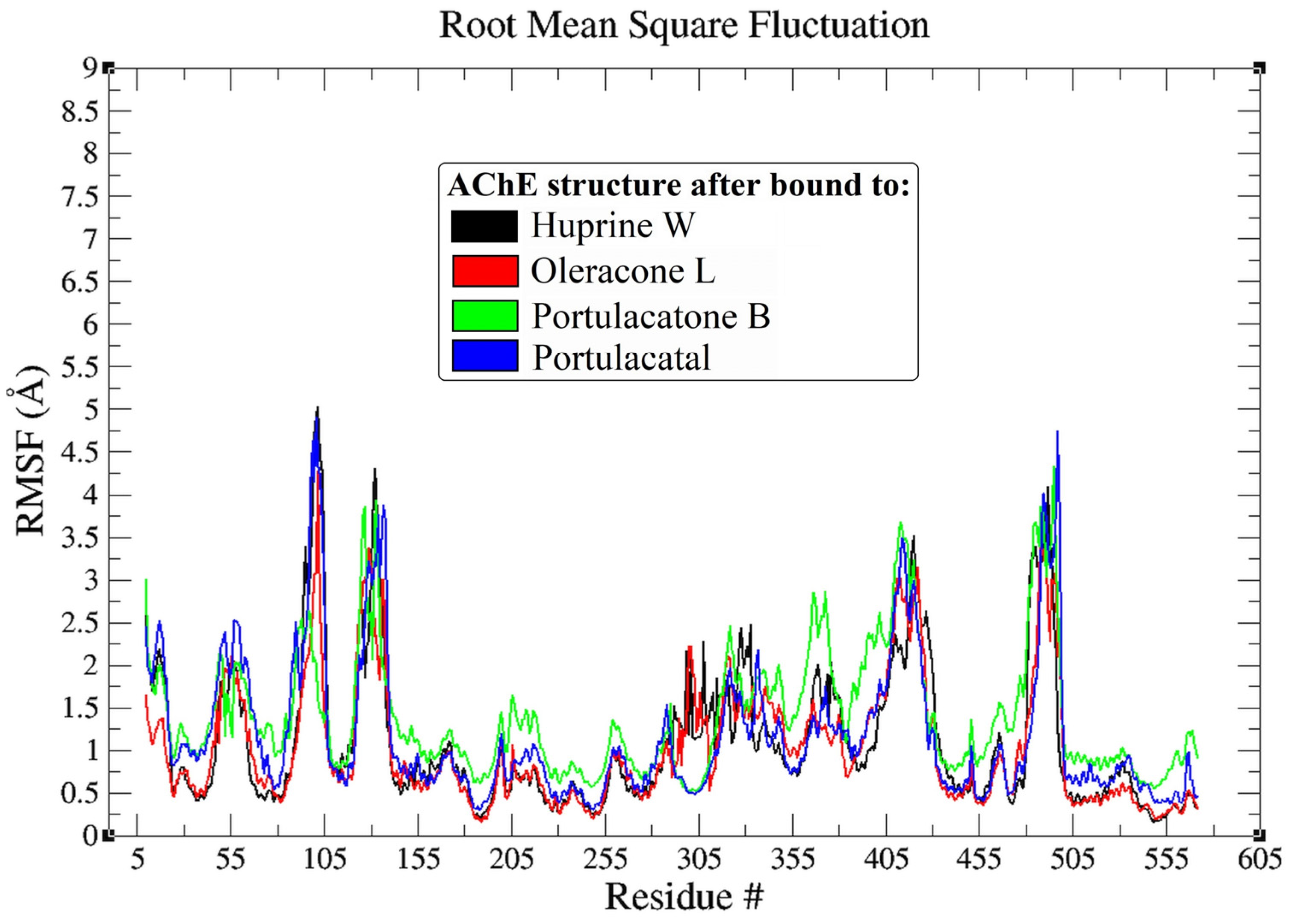
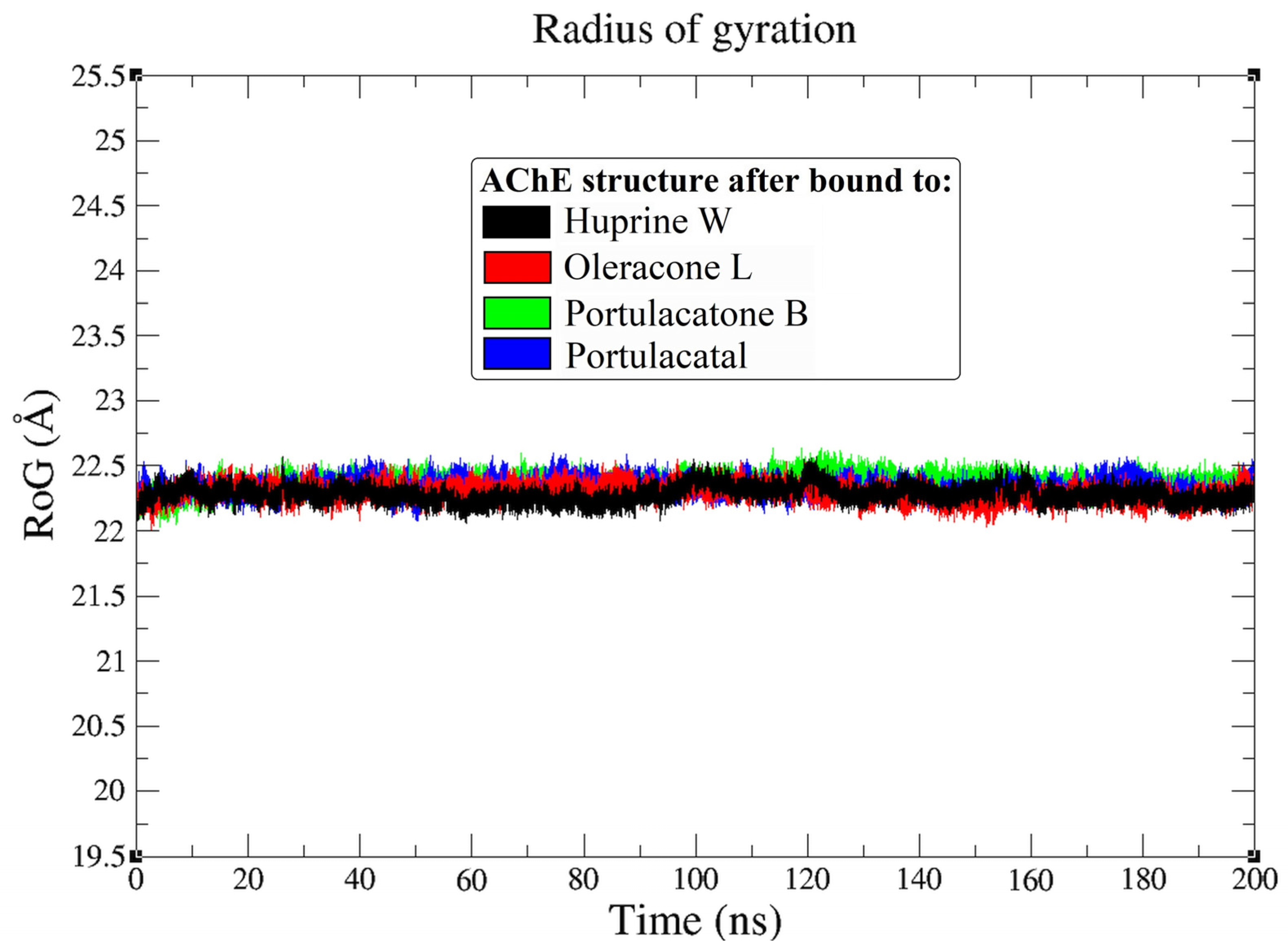
3.3. Molecular Dynamic Simulation
4. Discussion
4.1. Drug Likeness and ADME Predictions
4.2. Molecular Docking Simulation
4.3. Molecular Dynamic Simulation
5. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amin, F.; Shamsi, A.; Asghar, M.N.; Khaki, P.S.S.; Khan, M.S.; Tabrez, S.; Zaidi, S.K.; Khan, W.; Bano, B. Alzheimer’s: A Progressive Brain Disease: Causes, Symptoms, and Prevention. In Biological, Diagnostic and Therapeutic Advances in Alzheimer’s Disease: Non-Pharmacological Therapies for Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 2019; pp. 31–51. [Google Scholar]
- Bhavani, K.; Kuriyakose, J.; Sindhuja, R.; Chandrashekhar, U.; Ashab, A. A review on neurodegenerative disorder--alzheimer’s disease. Drug Invent. Today 2020, 13, 954. [Google Scholar]
- Alhawarri, M.B.; Al-Thiabat, M.G.; Dubey, A.; Tufail, A.; Fouad, D.; Alrimawi, B.H.; Dayoob, M. ADME profiling, molecular docking, DFT, and MEP analysis reveal cissamaline, cissamanine, and cissamdine from Cissampelos capensis Lf as potential anti-Alzheimer’s agents. RSC Adv. 2024, 14, 9878–9891. [Google Scholar] [CrossRef]
- Alhawarri, M.B.; Dianita, R.; Rawa, M.S.A.; Nogawa, T.; Wahab, H.A. Potential Anti-Cholinesterase Activity of Bioactive Compounds Extracted from Cassia grandis Lf and Cassia timoriensis DC. Plants 2023, 12, 344. [Google Scholar] [CrossRef]
- Alhawarri, M.B.; Dianita, R.; Razak, K.N.A.; Mohamad, S.; Nogawa, T.; Wahab, H.A. Antioxidant, anti-inflammatory, and inhibition of acetylcholinesterase potentials of Cassia timoriensis DC. flowers. Molecules 2021, 26, 2594. [Google Scholar] [CrossRef]
- Moreira, N.C.d.S.; Lima, J.E.B.d.F.; Marchiori, M.F.; Carvalho, I.; Sakamoto-Hojo, E.T. Neuroprotective effects of cholinesterase inhibitors: Current scenario in therapies for Alzheimer’s disease and future perspectives. J. Alzheimer’s Dis. Rep. 2022, 6, 177–193. [Google Scholar] [CrossRef]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The state of the art on acetylcholinesterase inhibitors in the treatment of Alzheimer’s disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029113. [Google Scholar] [CrossRef]
- Ozdemir, Z.; Ozcelik, A.; Uysal, M. Approaches based on cholinergic hypothesis and cholinesterase inhibitors in the treatment of alzheimer’s disease. Front. Clin. Drug Res.-Alzheimer Disord. 2019, 8, 154–190. [Google Scholar]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Begum, M.; Thangapandiyan, S.; Rahman, M.S.; Aleya, L.; Mathew, B.; Ahmed, M.; Barreto, G.E.; Ashraf, G.M. Cholinesterase inhibitors for Alzheimer’s disease: Multitargeting strategy based on anti-Alzheimer’s drugs repositioning. Curr. Pharm. Des. 2019, 25, 3519–3535. [Google Scholar] [CrossRef]
- Li, W.; Rang, Y.; Liu, H.; Liu, C. Update on new trends and progress of natural active ingredients in the intervention of Alzheimer’s disease, based on understanding of traditional Chinese and Western relevant theories: A review. Phytother. Res. 2023, 37, 3744–3764. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.H.; Kim, B.; Yang, G.; Kim, J.U. Natural products as the potential to improve Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2023, 24, 8827. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Guo, H.-Y.; Quan, Z.-S.; Shen, Q.-K.; Cui, H.; Li, X. Research progress of natural products and their derivatives against Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2171026. [Google Scholar] [CrossRef]
- Deng, M.; Yan, W.; Gu, Z.; Li, Y.; Chen, L.; He, B. Anti-neuroinflammatory potential of natural products in the treatment of Alzheimer’s disease. Molecules 2023, 28, 1486. [Google Scholar] [CrossRef]
- Puri, V.; Kanojia, N.; Sharma, A.; Huanbutta, K.; Dheer, D.; Sangnim, T. Natural product-based pharmacological studies for neurological disorders. Front. Pharmacol. 2022, 13, 1011740. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Alajmi, M.F.; Alsaweed, M.; Jamal, Q.M.S.; Alasiry, S.M.; Albaker, A.B.; Hamed, M.; Kamal, M.; Albadrani, H.M. Multitargeted virtual screening and molecular simulation of natural product-like compounds against GSK3β, NMDA-receptor, and BACE-1 for the management of Alzheimer’s disease. Pharmaceuticals 2023, 16, 622. [Google Scholar] [CrossRef] [PubMed]
- Tung, B.T.; Hue, N.T.; Long, N.V.; Ngoc, N.T. Medicinal Herbs Against Central Nervous System Disorders. In Pharmacological Benefits of Natural Agents; IGI Global: Hershey, PA, USA, 2023; pp. 85–103. [Google Scholar]
- Awuchi, C.G. Plants, phytochemicals, and natural practices in complementary and alternative system of medicine for treatment of central nervous system disorders. Int. J. Food Prop. 2023, 26, 1190–1213. [Google Scholar] [CrossRef]
- Islam, A.; Mishra, A.; Ahsan, R.; Fareha, S. Phytopharmaceuticals and Herbal Approaches to Target Neurodegenerative Disorders. Drug Res. 2023, 73, 388–407. [Google Scholar] [CrossRef]
- Srivastava, A.; Srivastava, P.; Pandey, A.; Khanna, V.; Pant, A. Phytomedicine: A potential alternative medicine in controlling neurological disorders. In New Look to Phytomedicine; Elsevier: Amsterdam, The Netherlands, 2019; pp. 625–655. [Google Scholar]
- Kakooza-Mwesige, A. The importance of botanical treatments in traditional societies and challenges in developing countries. Epilepsy Behav. 2015, 52, 297–307. [Google Scholar] [CrossRef]
- Perera, P.K.; Dahanayake, J.M.; Diddeniya, J.I.D. Pharmacology, Toxicology, and Therapeutic Effects of Metals and Minerals Used in Traditional Medicine. In Medical Geology: En Route to One Health; John and Wiley and Sons: Hoboken, NJ, USA, 2023; pp. 303–313. [Google Scholar]
- Zhou, Y.-X.; Xin, H.-L.; Rahman, K.; Wang, S.-J.; Peng, C.; Zhang, H. Portulaca oleracea L.: A review of phytochemistry and pharmacological effects. BioMed Res. Int. 2015, 2015, 925631. [Google Scholar] [CrossRef]
- Iranshahy, M.; Javadi, B.; Iranshahi, M.; Jahanbakhsh, S.P.; Mahyari, S.; Hassani, F.V.; Karimi, G. A review of traditional uses, phytochemistry and pharmacology of Portulaca oleracea L. J. Ethnopharmacol. 2017, 205, 158–172. [Google Scholar] [CrossRef]
- Montoya-García, C.O.; García-Mateos, R.; Becerra-Martínez, E.; Toledo-Aguilar, R.; Volke-Haller, V.H.; Magdaleno-Villar, J.J. Bioactive compounds of purslane (Portulaca oleracea L.) according to the production system: A review. Sci. Hortic. 2023, 308, 111584. [Google Scholar] [CrossRef]
- Masoodi, M.H.; Ahmad, B.; Mir, S.R.; Zargar, B.A.; Tabasum, N. Portulaca oleracea L. a review. J. Pharm. Res. 2011, 4, 3044–3048. [Google Scholar]
- Cui, X.; Ying, Z.; Ying, X.; Jia, L.; Yang, G. Three new alkaloids from Portulaca oleracea L. and their bioactivities. Fitoterapia 2021, 154, 105020. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, W.; Chen, X.; Xiao, J. Inhibition of flavonoids on acetylcholine esterase: Binding and structure–activity relationship. Food Funct. 2014, 5, 2582–2589. [Google Scholar] [CrossRef]
- Tanoli, S.T.; Ramzan, M.; Hassan, A.; Sadiq, A.; Jan, M.S.; Khan, F.A.; Ullah, F.; Ahmad, H.; Bibi, M.; Mahmood, T. Design, synthesis and bioevaluation of tricyclic fused ring system as dual binding site acetylcholinesterase inhibitors. Bioorganic Chem. 2019, 83, 336–347. [Google Scholar] [CrossRef]
- Silvia Matos, K.; FF da Cunha, E.; Abagyan, R.; C Ramalho, T. Computational evidence for the reactivation process of human acetylcholinesterase inhibited by carbamates. Comb. Chem. High Throughput Screen. 2014, 17, 554–564. [Google Scholar] [CrossRef]
- Carmo Carreiras, M.; Mendes, E.; Jesus Perry, M.; Paula Francisco, A.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- Jankowska, A.; Wesolowska, A.; Pawlowski, M.; Chlon-Rzepa, G. Multi-target-directed ligands affecting serotonergic neurotransmission for Alzheimer’s disease therapy: Advances in chemical and biological research. Curr. Med. Chem. 2018, 25, 2045–2067. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl-and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef]
- Tariq, S.; Mutahir, S.; Khan, M.A.; Mutahir, Z.; Hussain, S.; Ashraf, M.; Bao, X.; Zhou, B.; Stark, C.B.; Khan, I.U. Synthesis, in vitro cholinesterase inhibition, molecular docking, DFT, and ADME studies of novel 1, 3, 4-oxadiazole-2-thiol derivatives. Chem. Biodivers. 2022, 19, e202200157. [Google Scholar] [CrossRef]
- Borges, N.M.; Sartori, G.R.; Ribeiro, J.F.; Rocha, J.R.; Martins, J.B.; Montanari, C.A.; Gargano, R. Similarity search combined with docking and molecular dynamics for novel hAChE inhibitor scaffolds. J. Mol. Model. 2018, 24, 41. [Google Scholar] [CrossRef]
- Khan, I.; Bakht, S.M.; Ibrar, A.; Abbas, S.; Hameed, S.; White, J.M.; Rana, U.A.; Zaib, S.; Shahid, M.; Iqbal, J. Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: Design, synthesis, X-ray diffraction analysis and molecular docking studies. RSC Adv. 2015, 5, 21249–21267. [Google Scholar] [CrossRef]
- Ejaz, S.A.; Fayyaz, A.; Mahmood, H.M.K.; Aziz, M.; Ejaz, S.R.; Alkhuriji, A.F.; Al-Megrin, W.A.I.; Aborode, A.T.; Batiha, G.E.-S. 4-Phthalimidobenzenesulfonamide Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: DFTs, 3D-QSAR, ADMET, and Molecular Dynamics Simulation. Neurodegener. Dis. 2023, 22, 122–138. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic. Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Yamari, I.; Abchir, O.; Mali, S.N.; Errougui, A.; Talbi, M.; El Kouali, M.; Chtita, S. The anti-SARS-CoV-2 activity of novel 9, 10-dihydrophenanthrene derivatives: An insight into molecular docking, ADMET analysis, and molecular dynamics simulation. Sci. Afr. 2023, 21, e01754. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach. Pharmaceuticals 2023, 16, 704. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. The Potential Anti-Cancerous Activity of Prunus amygdalus var. amara Extract. Processes 2023, 11, 1277. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Al-Thiabat, M.G.; Saqallah, F.G.; Gazzali, A.M.; Mohtar, N.; Yap, B.K.; Choong, Y.S.; Wahab, H.A. Heterocyclic substitutions greatly improve affinity and stability of folic acid towards FRα. An in silico insight. Molecules 2021, 26, 1079. [Google Scholar] [CrossRef]
- Alidmat, M.M.; Khairuddean, M.; Kamal, N.N.S.N.M.; Muhammad, M.; Wahab, H.A.; Althiabat, M.G.; Alhawarri, M.B. Synthesis, Characterization, Molecular Docking and Cytotoxicity Evaluation of New Thienyl Chalcone Derivatives against Breast Cancer Cells. Syst. Rev. Pharm. 2022, 13, 1. [Google Scholar]
- Alhawarri, M.B.; Olimat, S. Potential Serotonin 5-HT2A Receptor Agonist of Psychoactive Components of Silene undulata Aiton: LC-MS/MS, ADMET, and Molecular Docking Studies. In Current Pharmaceutical Biotechnology; Bentham Science Publishers: Sharjah, United Arab Emirates, 2024. [Google Scholar]
- Yunos, N.M.; Al-Thiabat, M.G.; Sallehudin, N.J. Quassinoids from Eurycoma longifolia as Potential Dihydrofolate Reductase Inhibitors: A Computational Study. In Current Pharmaceutical Biotechnology; Bentham Science Publishers: Sharjah, United Arab Emirates, 2024. [Google Scholar]
- Forli, W.; Halliday, S.; Belew, R.; Olson, A.J. AutoDock Version 4.2. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef]
- Ross, B.J. A Lamarckian evolution strategy for genetic algorithms. In Practical Handbook of Genetic Algorithms; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–16. [Google Scholar]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Rühle, V. Pressure Coupling/Barostats. Journal Club. 2008; pp. 1–5. Available online: https://www2.mpip-mainz.mpg.de/~andrienk/journal_club/barostats.pdf (accessed on 29 April 2024).
- Berendsen, H.J.; Postma, J.v.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.-H.; Gohlke, H. Efficient approximation of ligand rotational and translational entropy changes upon binding for use in MM-PBSA calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic interactions are a key to MDM2 inhibition by polyphenols as revealed by molecular dynamics simulations and MM/PBSA free energy calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov. 2012, 7, 99–107. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Luty, B.A.; Cheung, P.P.; Litman, A.H.; Owen, R.M.; Rose, P.W. The Pfizer Crystal Structure Database: An essential tool for structure-based design at Pfizer. J. Comput. Chem. 2022, 43, 1053–1062. [Google Scholar] [CrossRef]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorganic Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, Y.; Yin, W.; Li, J.; Wang, W.; Bai, F.; Xu, S.; Gong, Q.; Peng, T.; Hong, Y. Kinetics-driven drug design strategy for next-generation acetylcholinesterase inhibitors to clinical candidate. J. Med. Chem. 2021, 64, 1844–1855. [Google Scholar] [CrossRef]
- Bortolami, M.; Rocco, D.; Messore, A.; Di Santo, R.; Costi, R.; Madia, V.N.; Scipione, L.; Pandolfi, F. Acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease–a patent review (2016–present). Expert Opin. Ther. Pat. 2021, 31, 399–420. [Google Scholar] [CrossRef]
- Mathew, B.; Parambi, D.G.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019, 352, 1900177. [Google Scholar] [CrossRef]
- Agu, P.; Afiukwa, C.; Orji, O.; Ezeh, E.; Ofoke, I.; Ogbu, C.; Ugwuja, E.; Aja, P. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Anwar, T.; Kumar, P.; Khan, A.U. Modern tools and techniques in computer-aided drug design. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–30. [Google Scholar]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Acúrcio, R.C.; Leonardo-Sousa, C.; García-Sosa, A.T.; Salvador, J.A.; Florindo, H.F.; Guedes, R.C. Structural insights and binding analysis for determining the molecular bases for programmed cell death protein ligand-1 inhibition. Medchemcomm 2019, 10, 1810–1818. [Google Scholar] [CrossRef]
- Alves, M.J.; Froufe, H.J.; Costa, A.F.; Santos, A.F.; Oliveira, L.G.; Osório, S.R.; Abreu, R.M.; Pintado, M.; Ferreira, I.C. Docking studies in target proteins involved in antibacterial action mechanisms: Extending the knowledge on standard antibiotics to antimicrobial mushroom compounds. Molecules 2014, 19, 1672–1684. [Google Scholar] [CrossRef]
- Amir Rawa, M.S.; Al-Thiabat, M.G.; Nogawa, T.; Futamura, Y.; Okano, A.; Wahab, H.A. Naturally Occurring 8ß, 13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves. Pharmaceuticals 2022, 15, 902. [Google Scholar] [CrossRef]
- Yunos, N.M.; Wahab, H.A.; Al-Thiabat, M.G.; Sallehudin, N.J.; Jauri, M.H. In Vitro and In Silico Analysis of the Anticancer Effects of Eurycomanone and Eurycomalactone from Eurycoma longifolia. Plants 2023, 12, 2827. [Google Scholar] [CrossRef]
- Larue, L.; Kenzhebayeva, B.; Al-Thiabat, M.G.; Jouan–Hureaux, V.; Mohd–Gazzali, A.; Wahab, H.A.; Boura, C.; Yeligbayeva, G.; Nakan, U.; Frochot, C. tLyp–1: A peptide suitable to target NRP–1 receptor. Bioorganic Chem. 2023, 130, 106200. [Google Scholar] [CrossRef]
- Chadha, N.; Tiwari, A.K.; Kumar, V.; Lal, S.; Milton, M.D.; Mishra, A.K. Oxime-dipeptides as anticholinesterase, reactivator of phosphonylated-serine of AChE catalytic triad: Probing the mechanistic insight by MM-GBSA, dynamics simulations and DFT analysis. J. Biomol. Struct. Dyn. 2015, 33, 978–990. [Google Scholar] [CrossRef]
- Almeida, M.C.; Resende, D.I.; da Costa, P.M.; Pinto, M.M.; Sousa, E. Tryptophan derived natural marine alkaloids and synthetic derivatives as promising antimicrobial agents. Eur. J. Med. Chem. 2021, 209, 112945. [Google Scholar] [CrossRef]
- Al-Thiabat; Gazzali, A.; Mohtar, N.; Murugaiyah, V.; Kamarulzaman, E.E.; Yap, B.K.; Rahman, N.A.; Othman, R.; Wahab, H.A. Conjugated β-cyclodextrin enhances the affinity of folic acid towards FRα: Molecular dynamics study. Molecules 2021, 26, 5304. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; C Rosales-Hernández, M.; E Mendieta-Wejebe, J.; Martínez-Archundia, M.; Correa Basurto, J. Current tools and methods in molecular dynamics (MD) simulations for drug design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef]
- Da Fonseca, A.M.; Caluaco, B.J.; Madureira, J.M.C.; Cabongo, S.Q.; Gaieta, E.M.; Djata, F.; Colares, R.P.; Neto, M.M.; Fernandes, C.F.C.; Marinho, G.S. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-bond, SASA and MMGBSA. Mol. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef]
- Janati-Fard, F.; Housaindokht, M.R.; Monhemi, H. Investigation of structural stability and enzymatic activity of glucose oxidase and its subunits. J. Mol. Catal. B Enzym. 2016, 134, 16–24. [Google Scholar] [CrossRef]
- Goodman, M.F. Hydrogen bonding revisited: Geometric selection as a principal determinant of DNA replication fidelity. Proc. Natl. Acad. Sci. USA 1997, 94, 10493–10495. [Google Scholar] [CrossRef]
- Chakraborty, S.; Minda, R.; Salaye, L.; Bhattacharjee, S.K.; Rao, B.J. Active site detection by spatial conformity and electrostatic analysis—Unravelling a proteolytic function in shrimp alkaline phosphatase. PLoS ONE 2011, 6, e28470. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Model Name | Predicted Value | Comment | ||
|---|---|---|---|---|---|
| Oleracone L | Portulacatone B | Portulacatal | |||
| Drug-Likeness | *Lipinski Rule | Accepted | Accepted | Accepted | Compounds satisfying the Golden Triangle and GSK rules may have more favourable ADMET profiles [59,60]. |
| *Pfizer Rule | Accepted | Accepted | Accepted | ||
| *Golden Triangle | Accepted | Accepted | Accepted | ||
| *GSK Rule | Accepted | Accepted | Accepted | ||
| Absorption | Papp (Caco-2 Permeability) cm/s | −5.123 | −4.82 | −4.93 | Caco-2 Permeability: High: >−5.15 cm/s; low: <−5.15 cm/s [59,60]. |
| MDCK Permeability cm/s | 7 × 10−6 | 14 × 10−6 | 9 × 10−6 | MDCK permeability (cm/s): Low: <2 × 10−6; Medium: 2–20 × 10−6; High: >20 × 10−6 [59,60]. | |
| HIA (Human Intestinal Absorption) | 0.022 | 0.041 | 0.031 | HIA: High: <0.30; Low: >0.30 [59,60]. | |
| Distribution | PPB (Plasma Protein Binding) % | 88.12 | 86.15 | 79.55 | PPB Optimal: <90%. Drugs with lower 90% protein-bound may have a high therapeutic index [59,60]. |
| BBB (Blood–Brain Barrier) | 0.39 | 0.51 | 0.139 | Cross BBB: ≥0.1; Cannot cross BBB: <0.1 [59,60]. | |
| VD (Volume Distribution) L/kg | 0.49 | 0.52 | 0.711 | VD Optimal: 0.04–20 L/kg [59,60]. | |
| Fu (The fraction unbound in plasms) % | 5.62 | 8.71 | 11.55 | Fu: Low: 5%; Moderate: 5~20%; High: >20% [59,60]. | |
| Metabolism | CYP1A2 inhibitor | Yes | Yes | Yes | |
| CYP2C19 inhibitor | No | No | No | ||
| CYP2C9 inhibitor | No | No | No | ||
| CYP2D6 inhibitor | No | No | No | ||
| CYP3A4 inhibitor | No | No | No | ||
| Excretion | *CL (Clearance Rate) mL/min/kg | 10.59 | 13.78 | 13.16 | CL: High: >15 mL/min/kg; Moderate: CL 5–15 mL/min/kg; Low: CL < 5 mL/min/kg [59,60]. |
| T ½ (Half Lifetime) h | 0.85 | 0.76 | 0.88 | T ½: Long half-life: > 3 h; Short half-life: < 3 h [59,60]. | |
| Compounds | Free Binding Energy (kcal/mol) | Molecular Interactions Analysis within the AChE Active Binding Site | |||
|---|---|---|---|---|---|
| H-Bond | Distance (Å) | Pi-Sigma | Hydrophobic Interaction | ||
| Oleracone L | −10.75 | ASN87, ASN87, GLY122, SER203, and HIS447 | 1.96, 2.51, 2.86, 2.04, and 1.89 | ASP74 and TRP439 | TRP86 and TYR337 |
| Portulacatone B | −8.89 | HIS447 | 1.68 and 1.89 | ---------------- | TRP86, TYR337, TRP439, and TYR449 |
| Portulacatal | −9.95 | GLY122, SER203, TRP439, HIS447, and HIS447 | 2.07, 2.30, 2.49, 1.75, and 1.82 | ---------------- | TRP86, TYR337, and TRP439 |
| Co-crystalized Ligand (huprine W, original pose) | −9.49 | GLY122 and SER203 | 2.96 and 2.33 | ---------------- | TRP86, TYR337, TRP439, MET443, PRO446, and TYR449 |
| System | ΔGbind (kcal/mol) | Electrostatic (kcal/mol) | Van der Waal (kcal/mol) | Polar Salvation (kcal/mol) | Non-Polar Salvation (kcal/mol) |
|---|---|---|---|---|---|
| AChE-Huprine W | −28.43 ± 0.11 | −17.22 ± 0.12 | −14.64 ± 0.14 | 18.28 ± 0.11 | −14.85 ± 0.12 |
| AChE-Oleracone L | −31.74 ± 0.14 | −19.42 ± 0.12 | −17.64 ± 0.13 | 19.40 ± 0.12 | −14.08 ± 0.13 |
| AChE-Portulacatone B | −25.21 ± 0.13 | −14.60 ± 0.11 | −14.66 ± 0.14 | 18.11 ± 0.12 | −14.06 ± 0.13 |
| AChE-Portulacatal | −29.85 ± 0.11 | −17.86 ± 0.12 | −16.75 ± 0.14 | 19.21 ± 0.13 | −14.45 ± 0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, S.O. Computational Insight of Oleracone L, Portulacatone B, and Portulacatal from Portulaca oleracea L. as Potential Anticholinesterase Inhibitors for Alzheimer’s. Processes 2024, 12, 1456. https://doi.org/10.3390/pr12071456
Alshammari SO. Computational Insight of Oleracone L, Portulacatone B, and Portulacatal from Portulaca oleracea L. as Potential Anticholinesterase Inhibitors for Alzheimer’s. Processes. 2024; 12(7):1456. https://doi.org/10.3390/pr12071456
Chicago/Turabian StyleAlshammari, Shifaa O. 2024. "Computational Insight of Oleracone L, Portulacatone B, and Portulacatal from Portulaca oleracea L. as Potential Anticholinesterase Inhibitors for Alzheimer’s" Processes 12, no. 7: 1456. https://doi.org/10.3390/pr12071456