

Optimization of Sample Preparation Procedure for Determination of Fat-Soluble Vitamins in Milk and Infant Food by HPLC Technique
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Extraction Methods
2.1.1. Solvent Extraction
2.1.2. Enzymatic Hydrolysis
2.1.3. Solid-Phase Extraction
2.1.4. Hot Saponification

{kind=link}
{kind=link}
| Procedure No. | Sample Preparation |
|---|---|
| 1 | Extraction with ultra-pure water, 4 N HCl, and hexane. |
| 2 | Extraction with ultra-pure water and hexane. |
| 3 | Sample digestion with lipase, extraction with ethanol/methanol (95:5 v/v), and addition of K2CO3 and hexane. |
| 4 | Extraction with ethanol and ultra-pure water. Extract purification by SPE HLB. Elution with 20% ethyl acetate in acetonitrile. |
| 5 | Extraction with ethanol and ultra-pure water. Extract purification by SPE C18. Elution with methanol. |
| 6 | Hot saponification. Extraction of unsaponifables with hexane/ethyl acetate (85:15 v/v). |
| 7 | Extraction with ethanol and hexane. |
2.2. Chemicals
2.3. Equipment
2.4. Chromatographic Conditions
2.5. Method Validation
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padh, H. Vitamins for Optimal Health. In Functional Foods; Goldberg, I., Ed.; Springer: Boston, MA, USA, 1994; p. 13. [Google Scholar] [CrossRef]
- Bender, D.A. Nutritional Biochemistry of the Vitamins, 2nd ed.; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Singh, P.; Kesharwani, R.K.; Keservani, R.K. Antioxidants and Vitamins: Roles in Cellular Function and Metabolism. In Sustained Energy for Enhanced Human Functions and Activity; Academic Press: Cambridge, MA, USA, 2017; pp. 385–407. [Google Scholar] [CrossRef]
- Rafeeq, H.; Ahmad, S.; Tareen, M.B.K.; Shahzad, K.A.; Bashir, A.; Jabeen, R.; Tariq, S.; Shehzadi, I. Biochemistry of Fat Soluble Vitamins, Sources, Biochemical Functions and Toxicity. Haya Saudi J. Life Sci. 2020, 5, 188–196. [Google Scholar] [CrossRef]
- Głąbska, D.; Kołota, A.; Lachowicz, K.; Skolmowska, D.; Stachoń, M.; Guzek, D. The Influence of Vitamin D Intake and Status on Mental Health in Children: A Systematic Review. Nutrients 2021, 13, 952. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Causes of Nutrition-related Public Health Problems of Preschool Children: Available Diet. J. Pediatr. Gastroenterol. Nutr. 2006, 43 (Suppl. S3), S8–S12. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Awasthi, A. Role of vitamin A in child health and nutrition. Clin. Epidemiol. Glob. Health 2020, 8, 1039–1042. [Google Scholar] [CrossRef]
- Torquato, P.; Marinelli, R.; Bartolini, D.; Galli, F. Vitamin E: Nutritional aspects. In Molecular Nutrition–Vitamins; Academic Press: Cambridge, MA, USA, 2020; pp. 447–485. [Google Scholar] [CrossRef]
- Kozioł-Kozakowska, A.; Maresz, K. The Impact of Vitamin K2 (Menaquinones) in Children’s Health and Diseases: A Review of the Literature. Children 2022, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Alturfan, A.A. A Guide to Vitamins and Their Effects on Diseases; Chapter 4—Water-soluble vitamins; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2023. [Google Scholar]
- Ofoedu, C.E.; Iwouno, J.O.; Ofoedu, E.O.; Ogueke, C.C.; Igwe, V.S.; Agunwah, I.M.; Ofoedum, A.F.; Chacha, J.S.; Muobike, O.P.; Agunbiade, A.O.; et al. Revisiting food-sourced vitamins for consumer diet and health needs: A perspective review. PeerJ 2021, 9, e11940. [Google Scholar] [CrossRef] [PubMed]
- Adamec, J.; Jannasch, A.; Huang, J.; Hohman, E.; Fleet, J.C.; Peacock, M.; Ferruzzi, M.G.; Martin, B.; Weaver, C.M. Development and optimization of an LC-MS/MS-based method for simultaneous quantification of vitamin D2, vitamin D3, 25-hydroxyvitamin D2 and 25-hydroxyvitamin D3. J. Sep. Sci. 2011, 34, 2011. [Google Scholar] [CrossRef] [PubMed]
- Ertugrul, S.; Yucel, C.; Sertoglu, E.; Ozkan, Y.; Ozgurtas, T. Development and optimization of simultaneous determination of fat-soluble vitamins by liquid chromatography-tandem mass spectrometry. Chem. Phys. Lipids 2020, 230, 104932. [Google Scholar] [CrossRef] [PubMed]
- Ryynänen, M.; Lampi, A.-M.; Salo-Väänänen, P.; Ollilainen, V.; Piironen, V. A small-scale sample preparation method with HPLC analysis for determination of tocopherols and tocotrienols in cereals. J. Food Compos. Anal. 2004, 17, 749–765. [Google Scholar] [CrossRef]
- Katsa, M.; Proestos, C.; Komaitis, E. Determination of Fat Soluble Vitamins A and E in Infant Formulas by HPLC-DAD. Curr. Res. Nutr. Food Sci. J. 2016, 4, 92–96. [Google Scholar] [CrossRef]
- Álvarez, R.; Vaz, B.; Gronemeyer, H.; de Lera, Á.R. Functions, Therapeutic Applications, and Synthesis of Retinoids and Carotenoids. Chem. Rev. 2014, 114, 1–125. [Google Scholar] [CrossRef]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Van Veldhuizen, P.; et al. Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Malloy, P.J.; Gross, C. Chapter 9—Vitamin D. In Osteoporosis, 2nd ed.; Academic Press: Cambridge, MA, USA, 2001; Volume 1, pp. 257–303. [Google Scholar]
- Dominguez, L.J.; Farruggia, M.; Veronese, N.; Barbagallo, M. Vitamin D Sources, Metabolism, and Deficiency: Available Compounds and Guidelines for Its Treatment. Metabolites 2021, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Masuno, H.; Yamamoto, K.; Wang, X.; Choi, M.; Ooizumi, H.; Shinki, T.; Yamada, S. Rational Design, Synthesis, and Biological Activity of Novel Conformationally Restricted Vitamin D Analogues. J. Med. Chem. 2002, 45, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Chodynski, M.; Corcoran, A.; Marcinkowska, E.; Brown, G.; Kutner, A. Double Point Modified Analogs of Vitamin D as Potent Activators of Vitamin D Receptor. Curr. Pharm. Des. 2015, 21, 1741–1763. [Google Scholar] [CrossRef]
- Prieto, P.; Pineda, M.; Aguilar, M. Spectrophotometric Quantitation of Antioxidant Capacity through the Formation of a Phosphomolybdenum Complex: Specific Application to the Determination of Vitamin E. Anal. Biochem. 1999, 269, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.; Salvadó, V. Determination of eight water- and fat-soluble vitamins in multi-vitamin pharmaceutical formulations by high-performance liquid chromatography. J. Chromatogr. A 2000, 870, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; Chua, S.C.; Ong, H.Y.; Ong, C.N. High-performance liquid chromatographic method for routine determination of vitamins A and E and β-carotene in plasma. J. Chromatogr. B Biomed. Sci. Appl. 1992, 581, 41–47. [Google Scholar] [CrossRef]
- Rodas Mendoza, B.; Morera Pons, S.; Castellote Bargalló, A.I.; López-Sabater, M.C. Rapid determination by reversed-phase high-performance liquid chromatography of Vitamins A and E in infant formulas. J. Chromatogr. A 2003, 1018, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Newman, P. Metabolism and cell biology of vitamin K. Thromb. Haemost. 2008, 100, 530–547. [Google Scholar] [CrossRef]
- Shearer, M.J.; Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res. 2014, 55, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Ravisankar, P.; Reddy, A.A.; Nagalakshmi, B.; Koushik, O.S.; Kumar, B.V.; Anvith, P.S. The Comprehensive Review on Fat Soluble Vitamins. IOSR J. Pharm. 2015, 5, 12–28. [Google Scholar]
- Nako, Y.; Tomomasa, T.; Morikawa, A. Risk of hypervitaminosis D from prolonged feeding of high vitamin D premature infant formula. Pediatr. Int. 2004, 46, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Croatian National Ordinance on Processed Cereal-Based Food and Baby Food for Infants and Young Children-OG 126/2013 (Pravilnik o Prerađenoj Hrani na Bazi žitarica i dječjoj Hrani za Dojenčad i Malu Djecu-NN 126/2013). Available online: https://narodne-novine.nn.hr/clanci/sluzbeni/2013_10_126_2741.html (accessed on 4 July 2024).
- Guidance Document for Competent Authorities for the Control of Compliance with EU Legislation. Health and Consumers Directorate-General. 2012. Available online: https://food.ec.europa.eu/system/files/2016-10/labelling_nutrition-vitamins_minerals-guidance_tolerances_1212_en.pdf (accessed on 4 July 2024).
- Perretti, G.; Marconi, O.; Montanari, L.; Fantozzi, P. Fat-soluble vitamin extraction by analytical supercritical carbon dioxide. J. Amer. Oil Chem. Soc. 2003, 80, 629–633. [Google Scholar] [CrossRef]
- Sazali, N.H.; Alshishani, A.; Saad, B.; Chew, K.Y.; Chong, M.M.; Miskam, M. Salting-out assisted liquid–liquid extraction coupled with high-performance liquid chromatography for the determination of vitamin D3 in milk samples. R Soc. Open Sci. 2019, 6, 190952. [Google Scholar] [CrossRef] [PubMed]
- Yueqing, X.; Zhang, L.; Yang, R.; Yu, X.; Yu, L.; Ma, F.; Li, H.; Wang, X.; Li, P. Extraction and Determination of Vitamin K1in Foodsby Ultrasound-Assisted Extraction, SPE, and LC-MS/MS. Molecules 2020, 25, 839–851. [Google Scholar]
- Fanali, C.; D’Orazio, G.; Fanali, G.A. Advanced analytical techniques for fat-soluble vitamin analysis. Trends Anal. Chem. 2017, 87, 82–97. [Google Scholar] [CrossRef]
- Mathiasson, L.; Turner, C.; Berg, H.; Dahlberg, L.; Theobald, A.; Anklam, E.; Ginn, R.; Sharman, M.; Ulberth, F.; Gabernig, R. Development of methods for the determination of vitamins A, E and β-carotene in processed foods based on supercritical fluid extraction: A collaborative study. Food Addit. Contam. 2002, 19, 632–646. [Google Scholar] [CrossRef]
- Turner, C.; Persson, M.; Mathiasson, L.; Adlercreutz, P. Lipase-catalyzed reactions in organic and supercritical solvents:application to fat-soluble vitamin determination in milk powder and infant formula. Enzym. Microb. Technol. 2001, 29, 111–121. [Google Scholar] [CrossRef]
- Katsa, M.; Papalouka, N.; Mavrogianni, T.; Papagiannopoulou, I.; Kostakis, M.; Proestos, C.; Thomaidis, N.S. Comparative Study for the Determination of Fat-Soluble Vitamins in Rice Cereal Baby Foods Using HPLC-DAD and UHPLC-APCI-MS/MS. Foods 2021, 10, 648. [Google Scholar] [CrossRef]
- Turner, C.; King, J.W.; Mathiasson, L. Supercritical fluid extraction and chromatography for fat-soluble vitamin analysis. J. Chrmatogr. A 2001, 936, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.J. Status of Methodology for the Determination of Fat-Soluble Vitamins in Foods, Dietary Supplements, and Vitamin Premixes. J. AOAC Int. 2007, 90, 897–910. [Google Scholar] [PubMed]

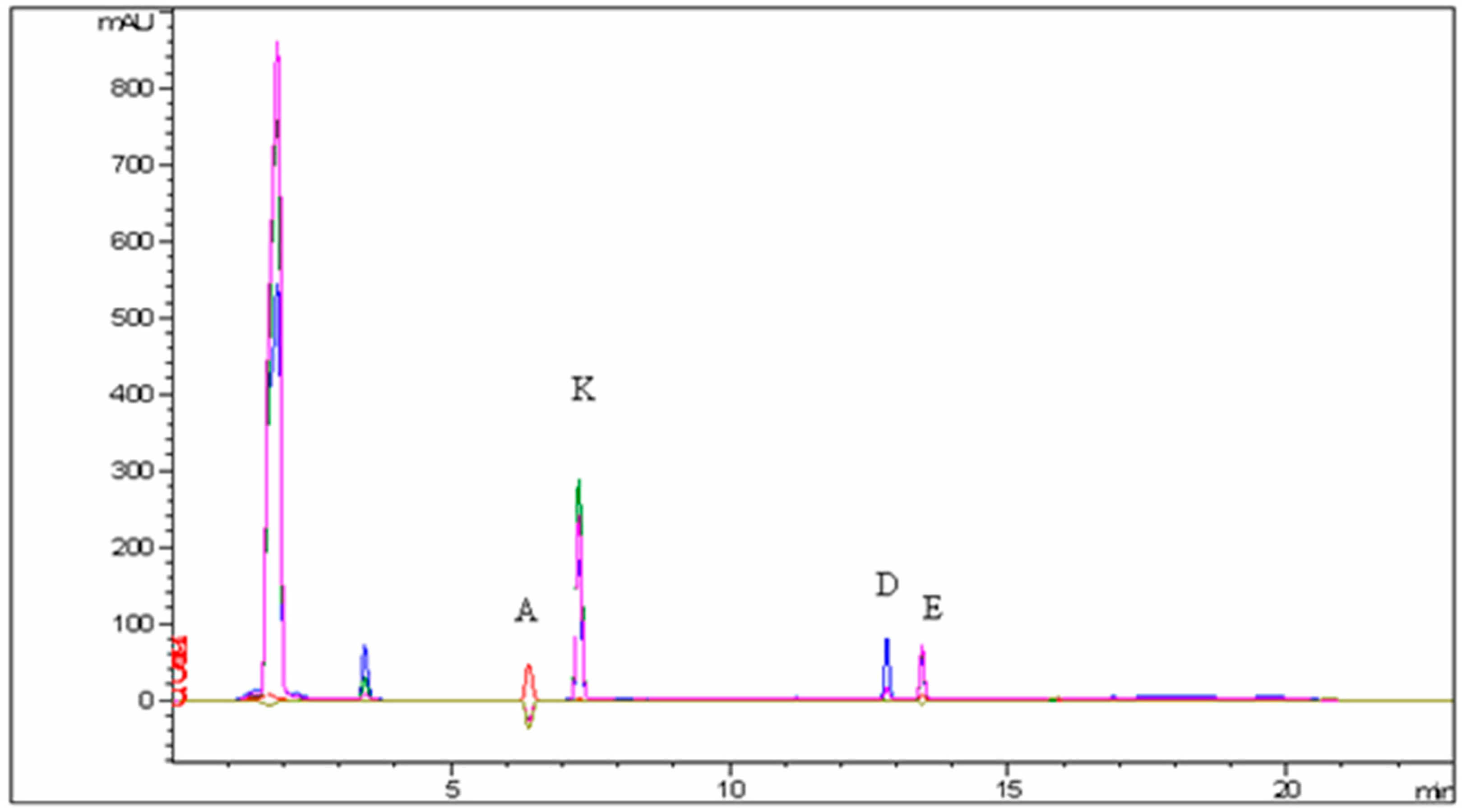
| Vitamin | Range (mg/mL) | Recovery (%) | Repeatability (%) | Intermediate Precision (%) | R2 | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|---|---|---|
| A | 0.072–28.900 | 97.4 | 1.521 | 2.896 | 0.999 | 0.010 | 0.100 |
| D | 0.048–19.200 | 96.1 | 1.325 | 3.250 | 0.999 | 0.010 | 0.100 |
| E | 0.013–27.000 | 98.3 | 1.021 | 2.128 | 1.000 | 0.010 | 0.100 |
| K | 0.013–25.213 | 96.2 | 1.102 | 3.012 | 0.999 | 0.010 | 0.100 |
| Vitamin | Order of Sample Preparation Efficiency |
|---|---|
| A | PR.5—PR.3—PR.6—PR.1—PR.7—PR.4—PR.2 |
| D | PR.5—PR.7—PR.4—PR.3—PR.6—PR.1—PR.2 |
| E | PR.5—PR.3—PR.6—PR.7—PR.4—PR.1—PR.2 |
| K | PR.5—PR.3—PR.2—PR.1—PR.4—PR.6—PR.7 |
| Test Statistic | Mean Rank | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Kruskal–Wallis H | df | Asymp. Sig. | P1 | P2 | P3 | P4 | P5 | P6 | ||
| Milk | Vitamin A | 90 | 83.536 | 5 | <0.01 | 38.93 | 8.00 | 64.40 | 23.00 | 82.60 | 56.07 |
| Vitamin D | 90 | 85.031 | 5 | <0.01 | 52.20 | 8.00 | 68.03 | 37.17 | 82.97 | 24.63 | |
| Vitamin E | 90 | 84.175 | 5 | <0.01 | 38.00 | 8.00 | 74.17 | 23.00 | 76.83 | 53.00 | |
| Vitamin K | 90 | 84.463 | 5 | <0.01 | 38.00 | 53.00 | 72.53 | 23.00 | 78.47 | 8.00 | |
| Cokolino | Vitamin A | 90 | 8.795 | 5 | 0.118 | 57.03 | 30.20 | 45.63 | 50.40 | 44.20 | 45.53 |
| Vitamin D | 90 | 10.402 | 5 | 0.065 | 33.77 | 46.17 | 51.13 | 35.93 | 46.53 | 59.47 | |
| Vitamin E | 90 | 84.111 | 5 | <0.01 | 83.00 | 20.60 | 51.00 | 10.40 | 68.00 | 40.00 | |
| Vitamin K | 90 | 3.283 | 5 | 0.656 | 35.87 | 51.03 | 46.30 | 46.30 | 43.73 | 49.77 | |
| Dry milk porridge | Vitamin A | 90 | 85.010 | 5 | <0.01 | 38.00 | 23.00 | 53.00 | 8.00 | 70.87 | 80.13 |
| Vitamin D | 90 | 86.341 | 5 | <0.01 | 38.33 | 8.00 | 83.00 | 23.00 | 68.00 | 52.67 | |
| Vitamin E | 90 | 86.546 | 5 | <0.01 | 83.00 | 8.00 | 53.00 | 38.00 | 68.00 | 23.00 | |
| Vitamin K | 90 | 43.230 | 5 | <0.01 | 62.43 | 9.07 | 46.17 | 61.47 | 43.67 | 50.20 | |
| Liquid milk porridge | Vitamin A | 90 | 84.163 | 5 | <0.01 | 83.00 | 8.00 | 44.43 | 23.00 | 46.57 | 68.00 |
| Vitamin D | 90 | 84.896 | 5 | <0.01 | 31.50 | 8.00 | 53.00 | 83.00 | 68.00 | 29.50 | |
| Vitamin E | 90 | 16.589 | 5 | <0.01 | 38.87 | 61.60 | 47.73 | 52.20 | 46.00 | 26.60 | |
| Vitamin K | 90 | 32.531 | 5 | <0.01 | 56.67 | 72.73 | 37.23 | 45.17 | 34.33 | 26.87 | |
| Sample Type | Vitamin A (µg/100 g) | ||||||
|---|---|---|---|---|---|---|---|
| Procedure 1 | Procedure 2 | Procedure 3 | Procedure 4 | Procedure 5 | Procedure 6 | Procedure 7 | |
| (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | |
| MILK | 352.33 ± 9.07 /460 (76.5%) | 13.75 ± 1.34 /460 (2.9%) | 454.33 ± 5.13/460 (98.7%) | 199.50 ± 2.12 /460 (43.3%) | 523.50 ± 30.41 /460 (114%) | 2.70 ± 0.10 /13 (21%) | 99.33 ± 2.33 /460 (22%) |
| COKOLINO | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| DRY MILK PORRIDGE | 218.33 ± 2.08 /375 (58.2%) | 32.40 ± 0.44 /375 (8.6%) | 279.67 ± 3.51 /375 (74.5%) | 13.15 ± 0.78 /375 (3.5%) | 40.67 ± 16.80 /375 (108%) | 2.00 ± 0.14 /13 (15.38%) | 111.67 ± 4.51 /375 (29.7%) |
| LIQUID MILK PORRIDGE | 92.27 ± 0.55 /65 (141.9%) | ND /65 | 59.00 ± 1.00 /65 (90%) | 3.87 ± 0.15 /65 (5.9%) | 59.67 ± 2.52 /65 (91.8%) | 76.40 ± 0.98 /65 (117.5%) | 24.33 ± 1.53 /65 (37.4%) |
| Sample Type | Vitamin D (µg/100 g) | ||||||
|---|---|---|---|---|---|---|---|
| Procedure 1 | Procedure 2 | Procedure 3 | Procedure 4 | Procedure 5 | Procedure 6 | Procedure 7 | |
| (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | |
| MILK | 4.00 ± 0.14 /13 (30.8%) | 0.30 ± 0.00 /13 (2.30%) | 9.27 ± 0.35 /13 (71.3%) | 3.30 ± 0.42 /13 (25.3%) | 11.50 ± 0.71 /13 (88.4%) | 2.70 ± 0.10 /13 (21%) | 4.63 ± 0.40 /13 (35.6%) |
| COKOLINO | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| DRY MILK PORRIDGE | 1.55 ± 0.21 /7.1 (21.8%) | ND /7.1 (0%) | 60.15 ± 0.07 /7.1 (847%) | 0.85 ± 0.07 /7.1 (11.9%) | 7.65 ± 0.35 /7.1 (108%) | 2.00 ± 0.14 /13 (15.38%) | 6.57 ± 0.31 /1.55 (212%) |
| LIQUID MILK PORRIDGE | 0.40 ± 0.00 /1.1 (36.6%) | ND /1.1 (0%) | 0.70 ± 0.00 /1.1 (63.6%) | 1.20 ± 0.10 1.1 (109%) | 0.83 ± 0.06 /1.1 (75.4%) | 0.40 ± 0.00 1.1 (36.3%) | 0.55 ± 0.07 /1.1 (50%) |
| Sample Type | Vitamin E (µg/100 g) | ||||||
|---|---|---|---|---|---|---|---|
| Procedure 1 | Procedure 2 | Procedure 3 | Procedure 4 | Procedure 5 | Procedure 6 | Procedure 7 | |
| (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | |
| MILK | 1.95 ± 0.21 /12 (16.2%) | 0.08 ± 0.04 /12 (0.66%) | 11.60 ± 0.62 /12 (96.6%) | 1.05 ± 0.07 /12 (8.75%) | 11.75 ± 0.49 /12 (97.9%) | 8.73 ± 0.45 /12 (72.7%) | 1.10 ± 0.00 /12 (0.09%) |
| COKOLINO | 6.50 ± 0.57 /3.3 (197%) | 0.80 ± 0.28 /3.3 (24.2%) | 3.27 ± 0.21 /33 (102%) | 0.54 ± 0.04 3.3 (16.3%) | 4.45 ± 0.49 /3.3 (135%) | 3.03 ± 0.21 /3.3 (91.8%) | 2.00 ± 0.10 /33 (6.1%) |
| DRY MILK PORRIDGE | 6.53 ± 0.32 /4.8 (136%) | 0.80 ± 0.14 /4.8 (167%) | 3.87 ± 0.06 /4.8 (80.6%) | 3.55 ± 0.07 /4.8 (73.9%) | 4.15 ± 0.07 /4.8 (86.4%) | 1.50 ± 0.14 /13 (11.5%) | 5.30 ± 0.00 /4.8 (110%) |
| LIQUID MILK PORRIDGE | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| Sample Type | Vitamin K (µg/100 g) | ||||||
|---|---|---|---|---|---|---|---|
| Procedure 1 | Procedure 2 | Procedure 3 | Procedure 4 | Procedure 5 | Procedure 6 | Procedure 7 | |
| (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | (MV ± SD /DV) (%) | |
| MILK | 17.00 ± 0.62 /38 (44.7%) | 21.00 ± 1.41 /38 (55.2%) | 36.33 ± 0.58 /38 (95%) | 5.80 ± 0.14 /38 (15.2%) | 37.50 ± 2.12 /38 (98.6%) | 2.05 ± 0.07 /38 (5.39%) | ND /38 (0%) |
| COKOLINO | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| DRY MILK PORRIDGE | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| LIQUID MILK PORRIDGE | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV | ND /no DV |
| Extraction Procedure | Solvents | Average Recoveries (%) | Time for Completion (h) | Temperature (°C) | Reliability of Results (Y/N) | Greenness Aspect (L/I/H) | Merits | Demerits |
|---|---|---|---|---|---|---|---|---|
| Saponification | citric acid ethanol KOH NaCl hexane/ethyl acetate (85:15 v/v) acetonitrile/methanol (75:25 v/v) | 20.3–88.1 | 1.5 | 70 | N | L | low digestion time | instability of vitamin K in an alkaline medium |
| Enzymatic hydrolysis | lipase phosphate buffer ethanol/methanol (95:5 v/v) K2CO3 hexsane acetonitrile/methanol (75:25 v/v) | 72.1–91.4 | 3 | 38 | Y | I | ability to break down complex molecules | time-consuming |
| Solvent extraction | 4 N HCl methanol ethanol hexane acetonitrile/methanol (75:25 v/v) | 70.3–90.1 | 1 | room | Y | I | easy to utilize | large amounts of solvents |
| Solid-phase extraction | ethanol methanol isopropanol/acetonitrile (50:50 v/v) 20% ethyl acetate acetonitrile/methanol (75:25 v/v) | 80.2–98.3 | 1.5 | room | Y | I | high recoveries | expensive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bošnir, J.; Bevardi, M.; Hećimović, I.; Budeč, M.; Juranović Cindrić, I.; Kober, R.; Jurak, G.; Lasić, D.; Brkić, D.; Racz, A. Optimization of Sample Preparation Procedure for Determination of Fat-Soluble Vitamins in Milk and Infant Food by HPLC Technique. Processes 2024, 12, 1530. https://doi.org/10.3390/pr12071530
Bošnir J, Bevardi M, Hećimović I, Budeč M, Juranović Cindrić I, Kober R, Jurak G, Lasić D, Brkić D, Racz A. Optimization of Sample Preparation Procedure for Determination of Fat-Soluble Vitamins in Milk and Infant Food by HPLC Technique. Processes. 2024; 12(7):1530. https://doi.org/10.3390/pr12071530
Chicago/Turabian StyleBošnir, Jasna, Martina Bevardi, Ida Hećimović, Maja Budeč, Iva Juranović Cindrić, Robert Kober, Gordana Jurak, Dario Lasić, Danijel Brkić, and Aleksandar Racz. 2024. "Optimization of Sample Preparation Procedure for Determination of Fat-Soluble Vitamins in Milk and Infant Food by HPLC Technique" Processes 12, no. 7: 1530. https://doi.org/10.3390/pr12071530