Seed Train Intensification and TFDF-Based Perfusion for MDCK Cell-Based Influenza A Virus Production


Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culturing
2.2. Influenza Virus Infection
2.3. Semi-Perfusion
2.4. TFDF-Based Perfusion
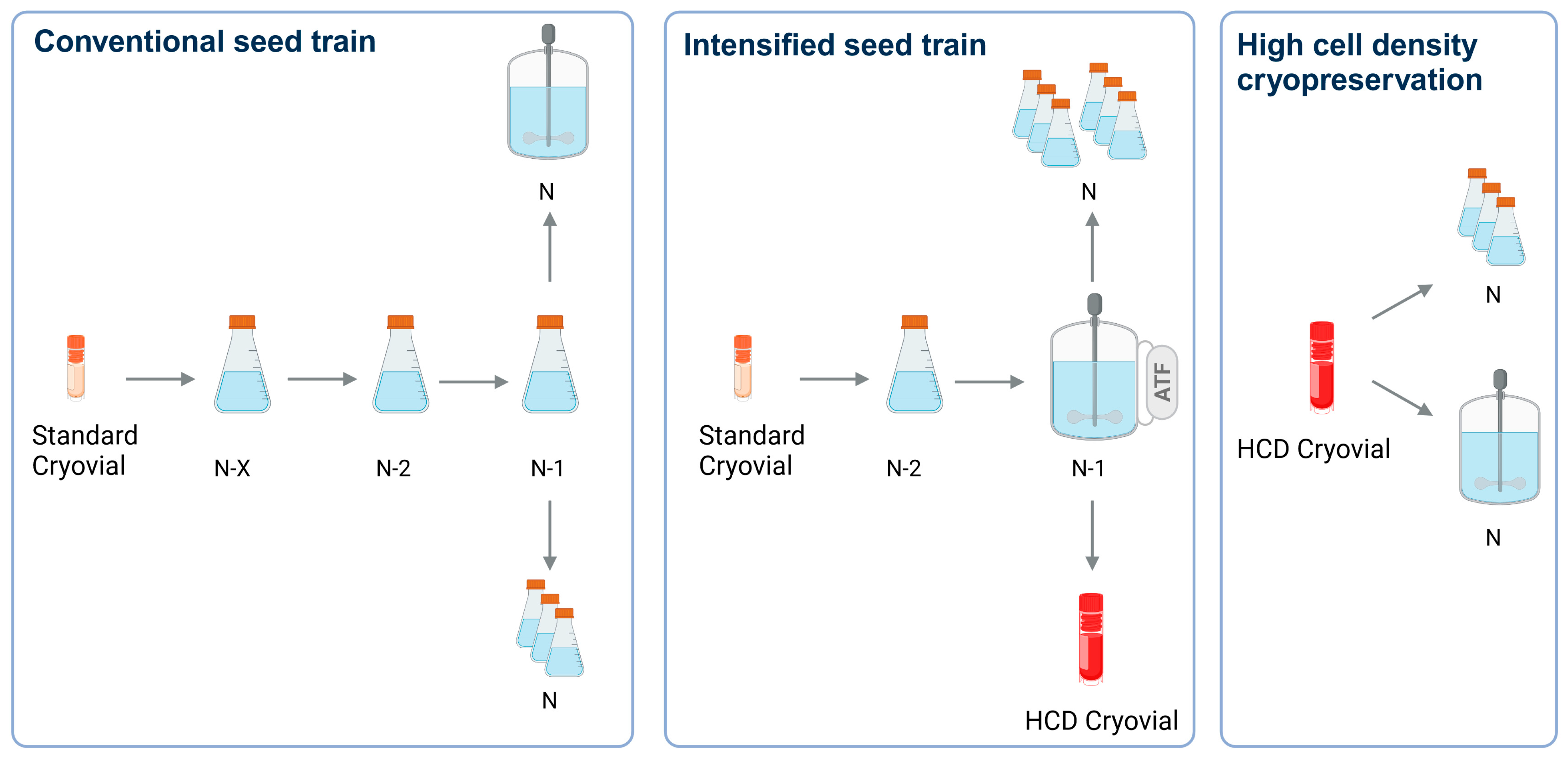
2.5. Seed Train Intensification via N-1 Perfusion
2.6. High Cell Density Cryopreservation for Direct Inoculation of a Production Process
2.7. Analytics
2.8. Statistical Analysis
3. Results
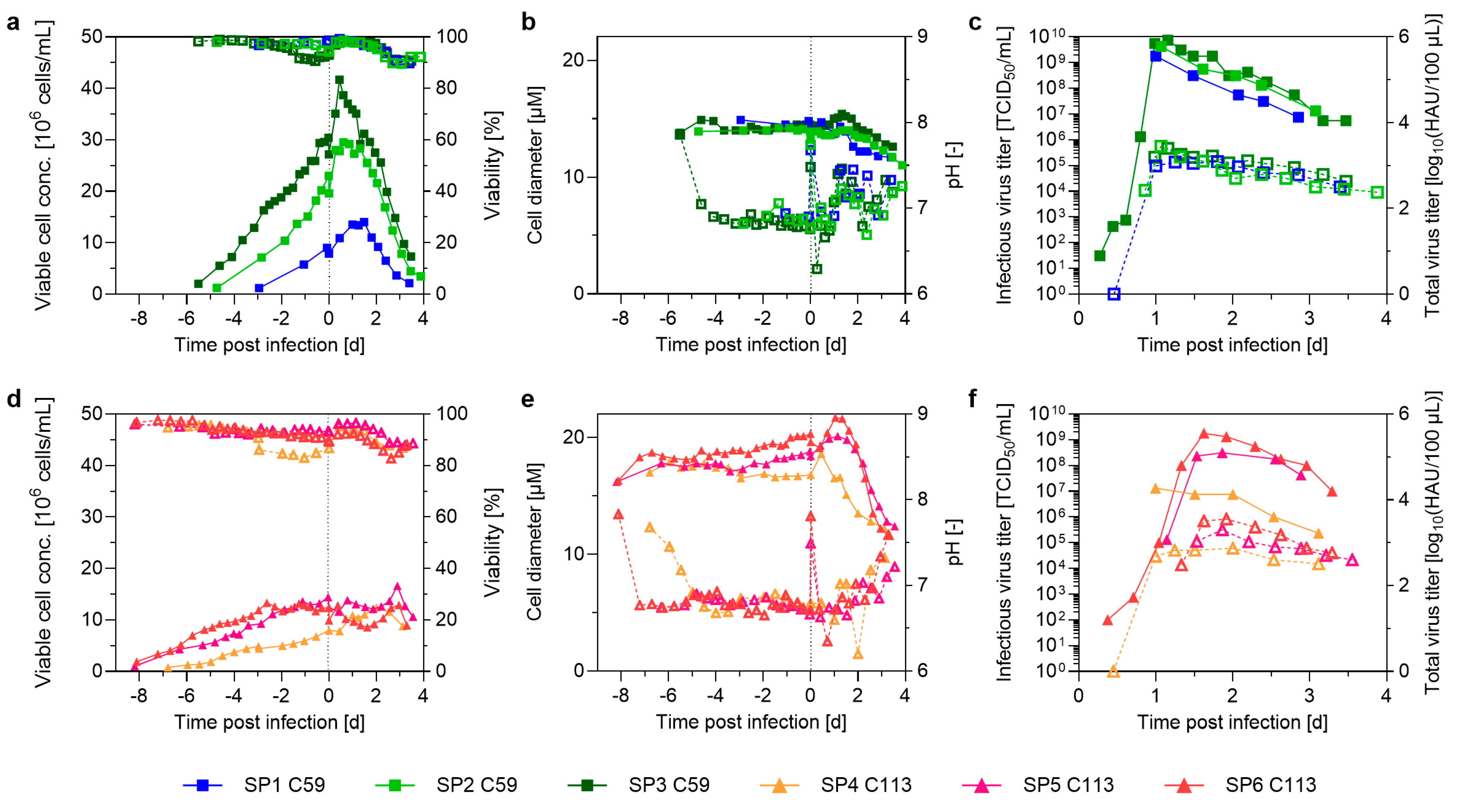
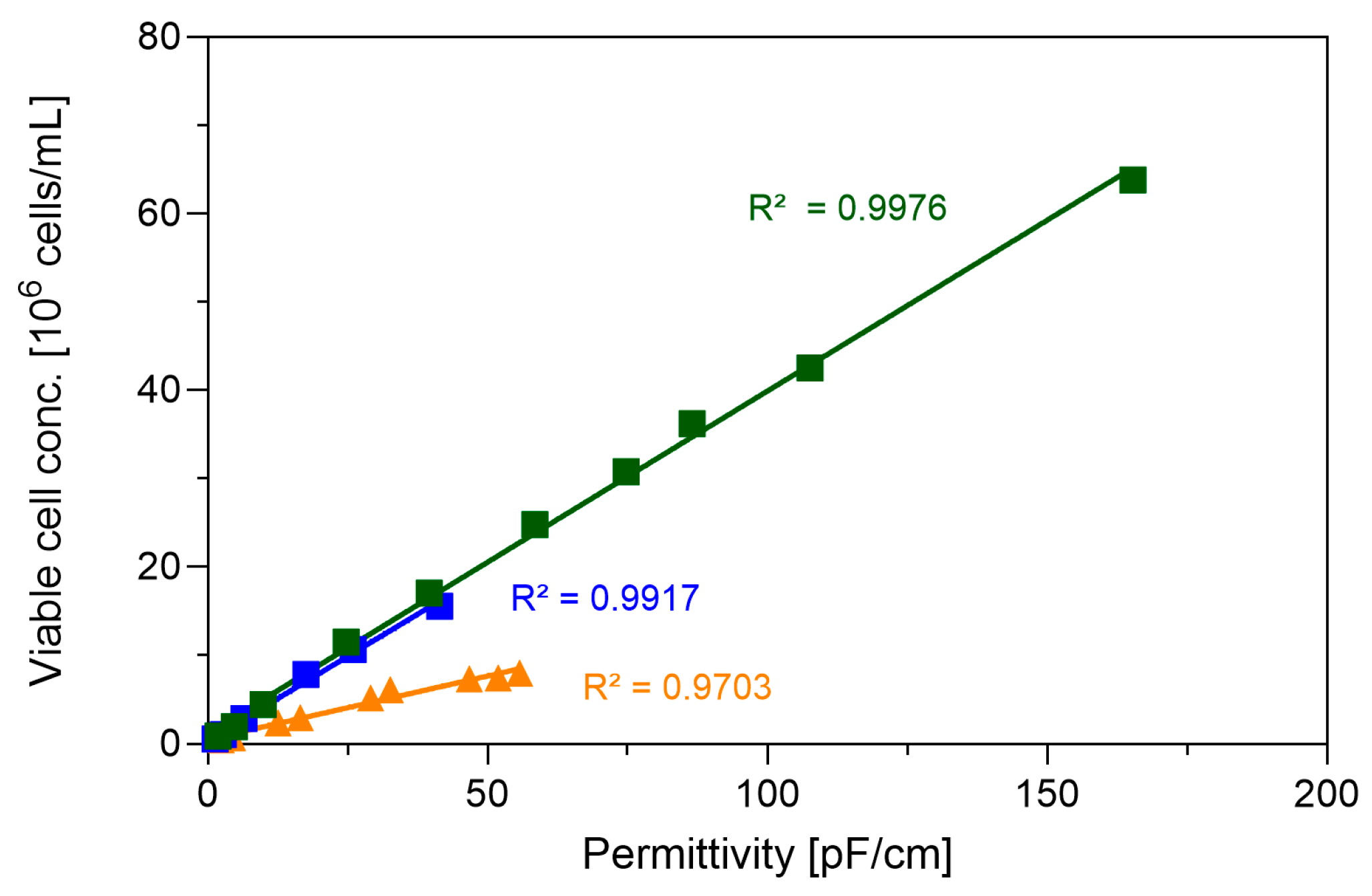
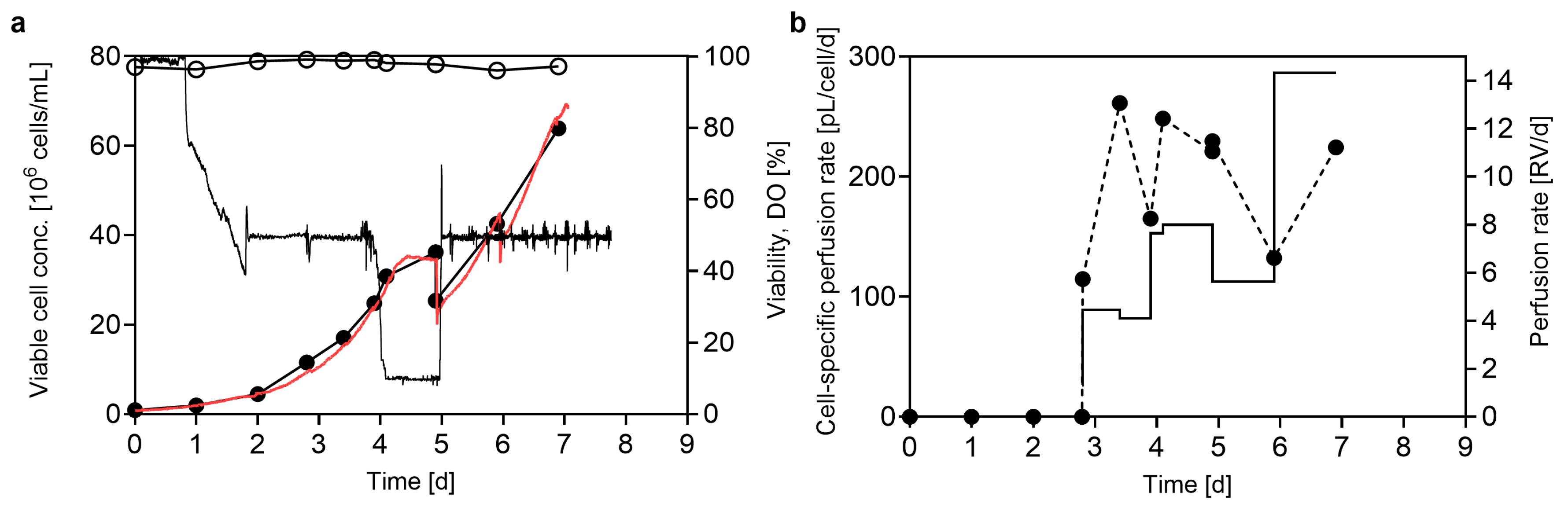
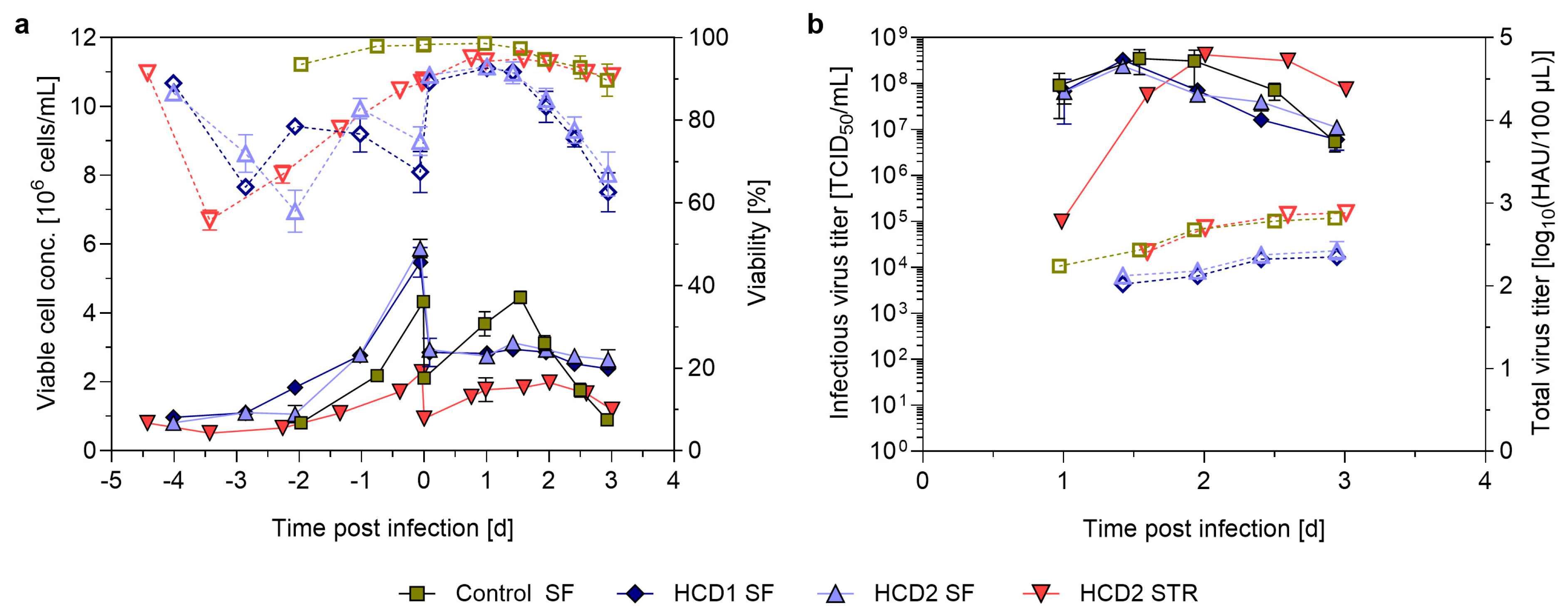
3.1. Pushing Towards High Cell Densities in a Small-Scale Model—Semi-Perfusion in Shake Flasks
3.2. TFDF-Based Perfusion Setup for IAV Production
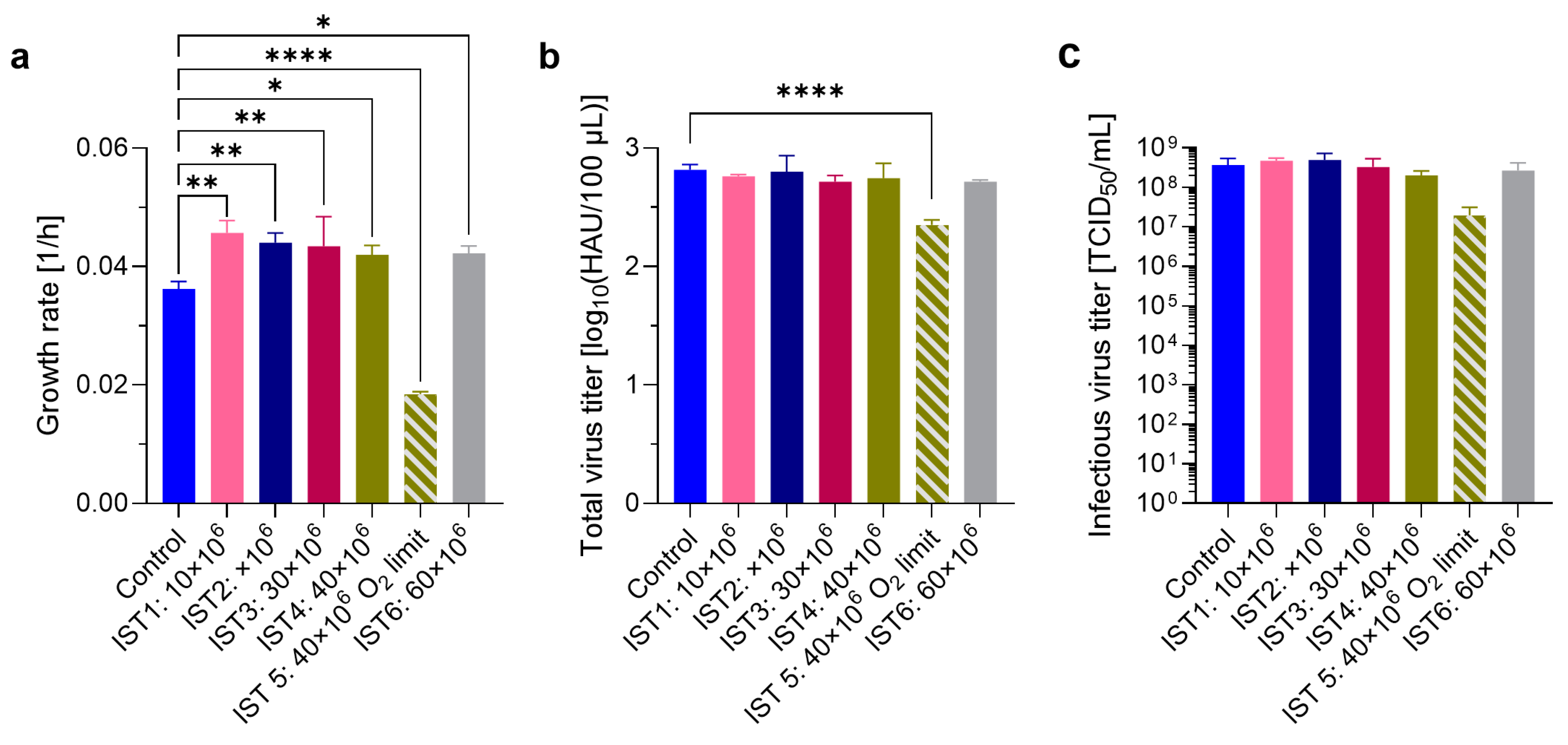
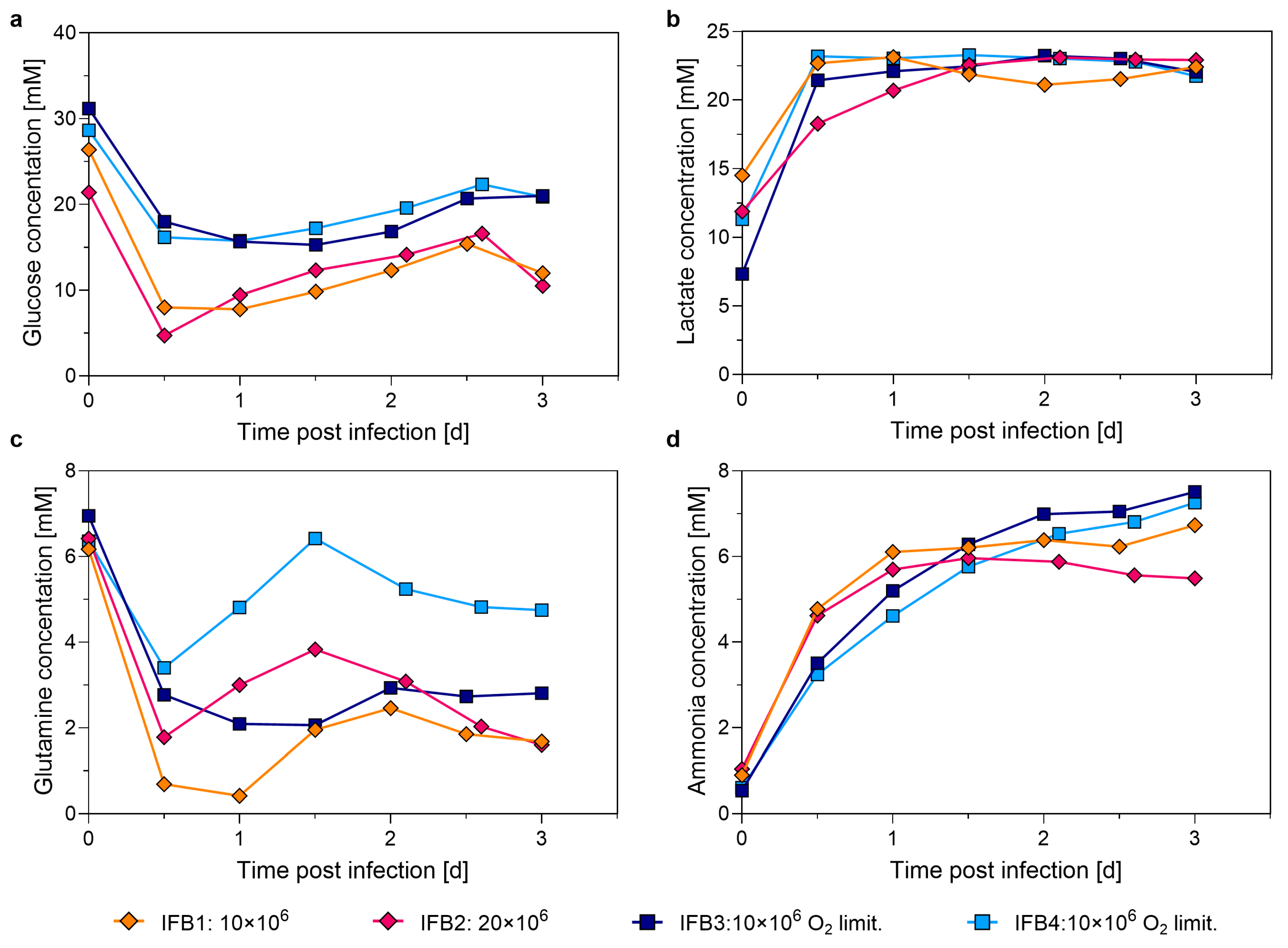
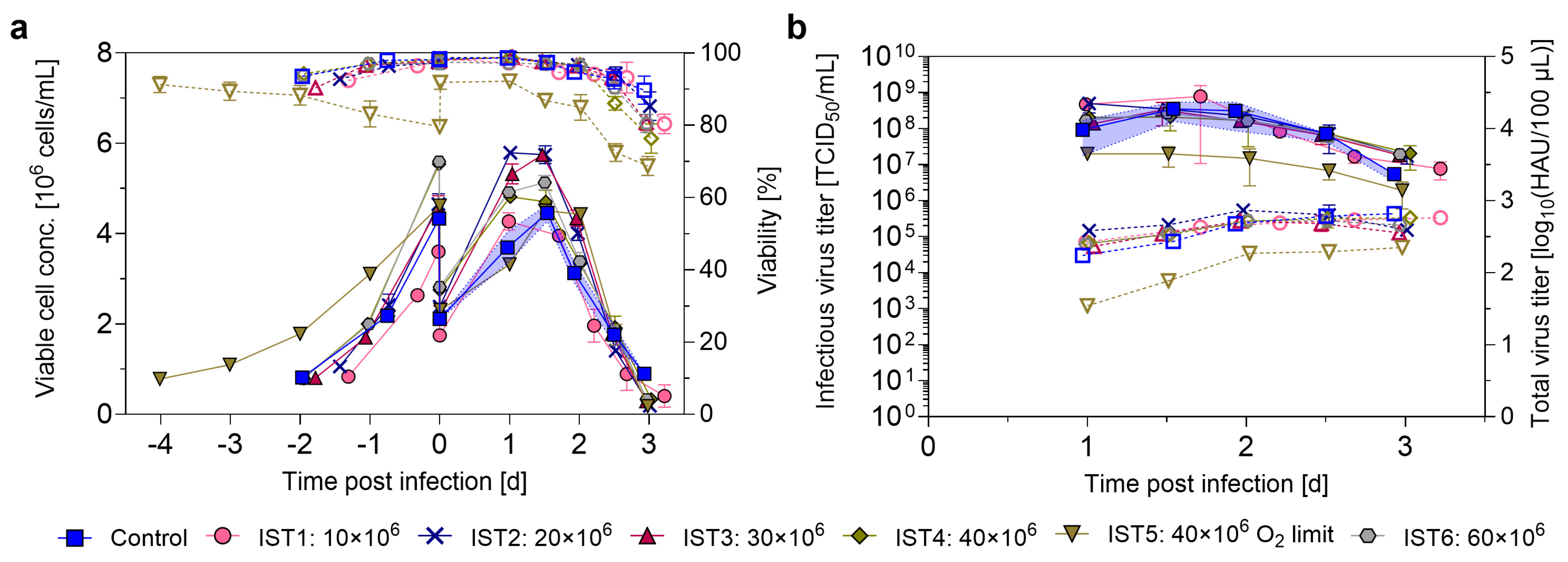
3.3. Intensifying the Seed Train by N-1 Perfusion
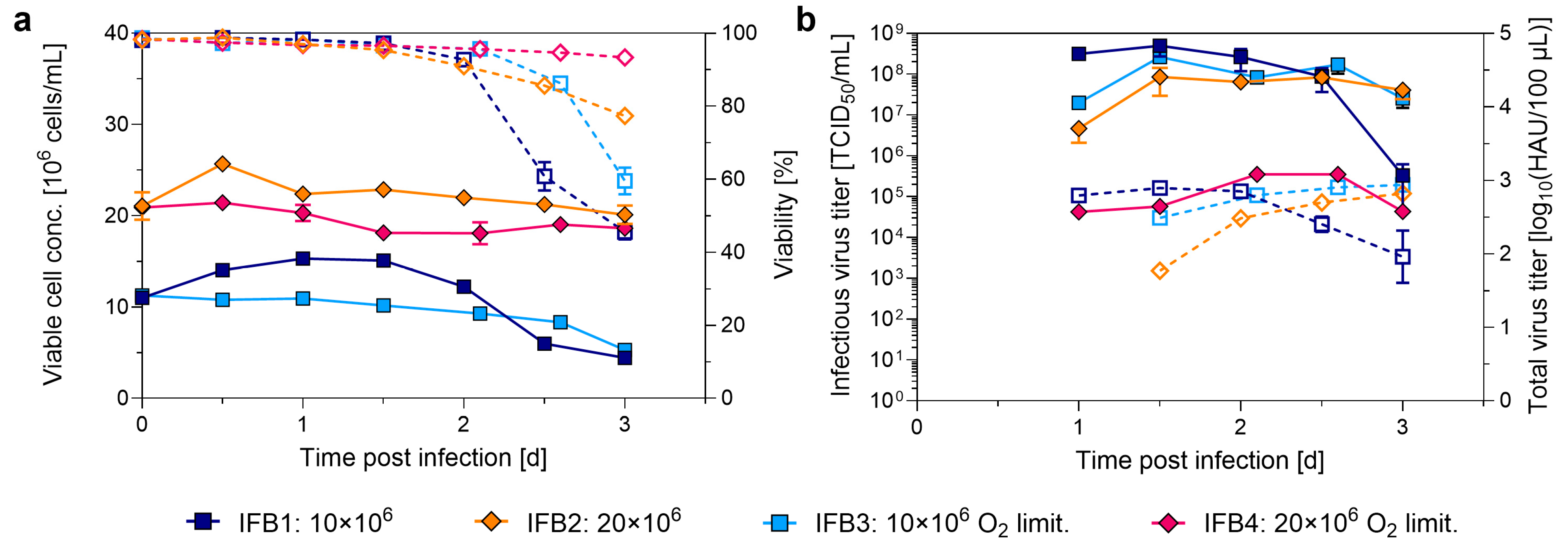
3.4. Testing the Impact of High Cell Density Cryopreservation on Cell Growth and IAV Production
4. Discussion
4.1. Reaching High Cell Densities in a Small-Scale Semi-Perfusion Model
4.2. Improved IAV Productivity Using a TFDF-Based Perfusion Setup
4.3. Evaluation of Seed Train Intensification by N-1 Perfusion
4.4. Implementation of High Cell Density Cryopreservation for IAV Production
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF | Alternating tangential flow filtration |
| B | Batch |
| CSPR | Cell-specific perfusion rate |
| CSVY | Cell-specific virus yield |
| DO | Dissolved oxygen |
| dpi | Days post-infection |
| HCD | High cell density |
| hpi | Hours post-infection |
| IAV | Influenza A virus |
| IFB | Intensified fed-batch |
| IST | Intensified seed train |
| MOI | Multiplicity of infection |
| PR | Perfusion rate |
| RV | Reactor volume |
| SP | Semi-perfusion |
| STR | Stirred-tank reactor |
| STY | Space-time yield |
| TFDF | Tangential flow depth filtration |
| TFF | Tangential flow filtration |
| TOI | Time of infection |
| VCC | Viable cell concentration |
| VVP | Volumetric virus productivity |
| wv | Working volume |
Appendix A

References
- Zinnecker, T.; Reichl, U.; Genzel, Y. Innovations in cell culture-based influenza vaccine manufacturing—From static cultures to high cell density cultivations. Hum. Vaccin. Immunother. 2024, 20, 2373521. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.; Vorlop, J. Animal Cells and Processes for the Replication of Influenza Viruses. CA2250714C, 6 November 2008. [Google Scholar]
- Gregersen, J.P.; Schmitt, H.J.; Trusheim, H.; Broker, M. Safety of MDCK cell culture-based influenza vaccines. Future Microbiol. 2011, 6, 143–152. [Google Scholar] [CrossRef]
- Pelz, L.; Göbel, S.; Jaen, K.; Reichl, U.; Genzel, Y. Upstream processing for viral vaccines—General aspects. In Bioprocessing of Viral Vaccines; Kamen, A., Cervera, L., Eds.; Taylor & Francis: Abingdon, UK, 2023; pp. 79–136. [Google Scholar]
- Göbel, S.; Pelz, L.; Reichl, U.; Genzel, Y. Upstream processing for viral vaccines—Process intensification. In Bioprocessing of Viral Vaccines; Kamen, A., Cervera, L., Eds.; Taylor & Francis: Abingdon, UK, 2023; pp. 137–174. [Google Scholar]
- Chotteau, V. Perfusion Processes. In Animal Cell Culture; Al-Rubeai, M., Ed.; Cell Engineering; Springer: Berlin/Heidelberg, Germany, 2015; pp. 407–443. [Google Scholar]
- Konstantinov, K.B.; Cooney, C.L. White Paper on Continuous Bioprocessing May 20–21 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Langer, E.S.; Rader, R.A. Continuous bioprocessing and perfusion: Wider adoption coming as bioprocessing matures. Bioprocess. J. 2014, 13, 50–55. [Google Scholar] [CrossRef]
- Genzel, Y.; Vogel, T.; Buck, J.; Behrendt, I.; Ramirez, D.V.; Schiedner, G.; Jordan, I.; Reichl, U. High cell density cultivations by alternating tangential flow (ATF) perfusion for influenza A virus production using suspension cells. Vaccine 2014, 32, 2770–2781. [Google Scholar] [CrossRef]
- Wu, Y.; Bissinger, T.; Genzel, Y.; Liu, X.; Reichl, U.; Tan, W.S. High cell density perfusion process for high yield of influenza A virus production using MDCK suspension cells. Appl. Microbiol. Biotechnol. 2021, 105, 1421–1434. [Google Scholar] [CrossRef]
- Vazquez-Ramirez, D.; Jordan, I.; Sandig, V.; Genzel, Y.; Reichl, U. High titer MVA and influenza A virus production using a hybrid fed-batch/perfusion strategy with an ATF system. Appl. Microbiol. Biotechnol. 2019, 103, 3025–3035. [Google Scholar] [CrossRef]
- Granicher, G.; Coronel, J.; Pralow, A.; Marichal-Gallardo, P.; Wolff, M.; Rapp, E.; Karlas, A.; Sandig, V.; Genzel, Y.; Reichl, U. Efficient influenza A virus production in high cell density using the novel porcine suspension cell line PBG.PK2.1. Vaccine 2019, 37, 7019–7028. [Google Scholar] [CrossRef]
- Coronel, J.; Behrendt, I.; Burgin, T.; Anderlei, T.; Sandig, V.; Reichl, U.; Genzel, Y. Influenza A virus production in a single-use orbital shaken bioreactor with ATF or TFF perfusion systems. Vaccine 2019, 37, 7011–7018. [Google Scholar] [CrossRef]
- Petiot, E.; Jacob, D.; Lanthier, S.; Lohr, V.; Ansorge, S.; Kamen, A.A. Metabolic and kinetic analyses of influenza production in perfusion HEK293 cell culture. BMC Biotechnol. 2011, 11, 84. [Google Scholar] [CrossRef]
- Coronel, J.; Granicher, G.; Sandig, V.; Noll, T.; Genzel, Y.; Reichl, U. Application of an Inclined Settler for Cell Culture-Based Influenza A Virus Production in Perfusion Mode. Front. Bioeng. Biotechnol. 2020, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Granicher, G.; Coronel, J.; Trampler, F.; Jordan, I.; Genzel, Y.; Reichl, U. Performance of an acoustic settler versus a hollow fiber-based ATF technology for influenza virus production in perfusion. Appl. Microbiol. Biotechnol. 2020, 104, 4877–4888. [Google Scholar] [CrossRef]
- Gobel, S.; Pelz, L.; Silva, C.A.T.; Bruhlmann, B.; Hill, C.; Altomonte, J.; Kamen, A.; Reichl, U.; Genzel, Y. Production of recombinant vesicular stomatitis virus-based vectors by tangential flow depth filtration. Appl. Microbiol. Biotechnol. 2024, 108, 240. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.A.T.; Kamen, A.A.; Henry, O. Intensified Influenza Virus Production in Suspension HEK293SF Cell Cultures Operated in Fed-Batch or Perfusion with Continuous Harvest. Vaccines 2023, 11, 1819. [Google Scholar] [CrossRef]
- Tran, M.Y.; Kamen, A.A. Production of Lentiviral Vectors Using a HEK-293 Producer Cell Line and Advanced Perfusion Processing. Front. Bioeng. Biotechnol. 2022, 10, 887716. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.P.; Fernandes, B.; Pineda, E.; Kudugunti, S.; Bransby, M.; Gantier, R.; Peixoto, C.; Alves, P.M.; Roldao, A.; Silva, R.J.S. AAV process intensification by perfusion bioreaction and integrated clarification. Front. Bioeng. Biotechnol. 2022, 10, 1020174. [Google Scholar] [CrossRef]
- Tao, Y.; Shih, J.; Sinacore, M.; Ryll, T.; Yusuf-Makagiansar, H. Development and implementation of a perfusion-based high cell density cell banking process. Biotechnol. Prog. 2011, 27, 824–829. [Google Scholar] [CrossRef]
- Müller, J.; Ott, V.; Eibl, D.; Eibl, R. Seed Train Intensification Using an Ultra-High Cell Density Cell Banking Process. Processes 2022, 10, 911. [Google Scholar] [CrossRef]
- Woodgate, J.M. Perfusion N-1 Culture—Opportunities for Process Intensification. In Biopharmaceutical Processing; ScienceDirect: Amsterdam, The Netherlands, 2018; pp. 755–768. [Google Scholar]
- Müller, J.; Ott, V.; Weiss, N.; Neubauer, P.; Eibl, D.; Eibl, R. Process Intensification Using a One-Step Inoculum Production and High-Seeded Fed-Batch Processes. Chem. Ing. Tech. 2022, 94, 1977–1984. [Google Scholar] [CrossRef]
- Jordan, M.; Mac Kinnon, N.; Monchois, V.; Stettler, M.; Broly, H. Intensification of large-scale cell culture processes. Curr. Opin. Chem. Eng. 2018, 22, 253–257. [Google Scholar] [CrossRef]
- Xu, J.; Rehmann, M.S.; Xu, M.; Zheng, S.; Hill, C.; He, Q.; Borys, M.C.; Li, Z.J. Development of an intensified fed-batch production platform with doubled titers using N-1 perfusion seed for cell culture manufacturing. Bioresour. Bioprocess. 2020, 7, 17. [Google Scholar] [CrossRef]
- Yongky, A.; Xu, J.; Tian, J.; Oliveira, C.; Zhao, J.; McFarland, K.; Borys, M.C.; Li, Z.J. Process intensification in fed-batch production bioreactors using non-perfusion seed cultures. MAbs 2019, 11, 1502–1514. [Google Scholar] [CrossRef]
- Stepper, L.; Filser, F.A.; Fischer, S.; Schaub, J.; Gorr, I.; Voges, R. Pre-stage perfusion and ultra-high seeding cell density in CHO fed-batch culture: A case study for process intensification guided by systems biotechnology. Bioprocess. Biosyst. Eng. 2020, 43, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Zinnecker, T.; Badri, N.; Araujo, D.; Thiele, K.; Reichl, U.; Genzel, Y. From single-cell cloning to high-yield influenza virus production—Implementing advanced technologies in vaccine process development. Eng. Life Sci. 2024, 24. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, T.; Fritsch, J.; Mihut, A.; Wu, Y.; Liu, X.; Genzel, Y.; Tan, W.S.; Reichl, U. Semi-perfusion cultures of suspension MDCK cells enable high cell concentrations and efficient influenza A virus production. Vaccine 2019, 37, 7003–7010. [Google Scholar] [CrossRef]
- Kalbfuss, B.; Knochlein, A.; Krober, T.; Reichl, U. Monitoring influenza virus content in vaccine production: Precise assays for the quantitation of hemagglutination and neuraminidase activity. Biologicals 2008, 36, 145–161. [Google Scholar] [CrossRef]
- Lavado-García, J.; Pérez-Rubio, P.; Cervera, L.; Gòdia, F. The cell density effect in animal cell-based bioprocessing: Questions, insights and perspectives. Biotechnol. Adv. 2022, 60, 108017. [Google Scholar] [CrossRef]
- Wohlpart, D.; Kirwan, D.; Gainer, J. Effects of cell density and glucose and glutamine levels on the respiration rates of hybridoma cells. Biotechnol. Bioeng. 1990, 36, 630–635. [Google Scholar] [CrossRef]
- Henry, O.; Dormond, E.; Perrier, M.; Kamen, A. Insights into adenoviral vector production kinetics in acoustic filter-based perfusion cultures. Biotechnol. Bioeng. 2004, 86, 765–774. [Google Scholar] [CrossRef]
- Vazquez-Ramirez, D.; Genzel, Y.; Jordan, I.; Sandig, V.; Reichl, U. High-cell-density cultivations to increase MVA virus production. Vaccine 2018, 36, 3124–3133. [Google Scholar] [CrossRef]
- Nikolay, A.; Bissinger, T.; Gränicher, G.; Wu, Y.; Genzel, Y.; Reichl, U. Perfusion Control for High Cell Density Cultivation and Viral Vaccine Production. In Animal Cell Biotechnology, 4th ed.; Pörtner, R., Ed.; Methods in Molecular Biology; Springer Protocols: Berlin/Heidelberg, Germany, 2020; pp. 141–168. [Google Scholar]
- Janoschek, S.; Schulze, M.; Zijlstra, G.; Greller, G.; Matuszczyk, J. A protocol to transfer a fed-batch platform process into semi-perfusion mode: The benefit of automated small-scale bioreactors compared to shake flasks as scale-down model. Biotechnol. Prog. 2019, 35, e2757. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Bidkhori, G.; Schwarz, H.; Malm, M.; Mebrahtu, A.; Field, R.; Sellick, C.; Hatton, D.; Varley, P.; Mardinoglu, A.; et al. Low Shear Stress Increases Recombinant Protein Production and High Shear Stress Increases Apoptosis in Human Cells. iScience 2020, 23, 101653. [Google Scholar] [CrossRef]
- Hein, M.D.; Chawla, A.; Cattaneo, M.; Kupke, S.Y.; Genzel, Y.; Reichl, U. Cell culture-based production of defective interfering influenza A virus particles in perfusion mode using an alternating tangential flow filtration system. Appl. Microbiol. Biotechnol. 2021, 105, 7251–7264. [Google Scholar] [CrossRef] [PubMed]
- Tona, R.M.; Shah, R.; Middaugh, K.; Steve, J.; Marques, J.; Roszell, B.R.; Jung, C. Process intensification for lentiviral vector manufacturing using tangential flow depth filtration. Mol. Ther. Methods Clin. Dev. 2023, 29, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.S.; Buchsteiner, M.; Gray, P.; Nielsen, L.K.; Quek, L.E. Dynamic metabolic flux analysis using B-splines to study the effects of temperature shift on CHO cell metabolism. Metab. Eng. Commun. 2015, 2, 46–57. [Google Scholar] [CrossRef]
- Martens, D.; Pan, X.; Wijffels, R.; Dalm, C. The relevance of cell size in a CHO fed batch process: Metabolic and transcriptomic characterization. In Proceedings of the Cell Culture Engineering XVI, Tampa, FL, USA, 6–11 May 2018. [Google Scholar]
- Kiehl, T.R.; Shen, D.; Khattak, S.F.; Jian Li, Z.; Sharfstein, S.T. Observations of cell size dynamics under osmotic stress. Cytom. A 2011, 79, 560–569. [Google Scholar] [CrossRef]
- Schellenberg, J.; Nagraik, T.; Wohlenberg, O.J.; Ruhl, S.; Bahnemann, J.; Scheper, T.; Solle, D. Stress-induced increase of monoclonal antibody production in CHO cells. Eng. Life Sci. 2022, 22, 427–436. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, W.; Han, P.; Zhang, J.; Zhu, Y.; Meng, X.; Zhang, J.; Hu, Y.; Yi, Z.; Wang, R. Influenza A virus (H1N1) triggers a hypoxic response by stabilizing hypoxia-inducible factor-1alpha via inhibition of proteasome. Virology 2019, 530, 51–58. [Google Scholar] [CrossRef]
- Schneider, M.; Marison, I.W.; von Stockar, U. The importance of ammonia in mammalian cell culture. J. Biotechnol. 1996, 46, 161–185. [Google Scholar] [CrossRef]
- Sieck, J.B.; Budach, W.E.; Suemeghy, Z.; Leist, C.; Villiger, T.K.; Morbidelli, M.; Soos, M. Adaptation for survival: Phenotype and transcriptome response of CHO cells to elevated stress induced by agitation and sparging. J. Biotechnol. 2014, 189, 94–103. [Google Scholar] [CrossRef]
- Godoy-Silva, R.; Chalmers, J.J.; Casnocha, S.A.; Bass, L.A.; Ma, N. Physiological responses of CHO cells to repetitive hydrodynamic stress. Biotechnol. Bioeng. 2009, 103, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Monteil, D.T.; Juvet, V.; Paz, J.; Moniatte, M.; Baldi, L.; Hacker, D.L.; Wurm, F.M. A comparison of orbitally-shaken and stirred-tank bioreactors: pH modulation and bioreactor type affect CHO cell growth and protein glycosylation. Biotechnol. Prog. 2016, 32, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.A.; Gibson, M.I. Post-Thaw Culture and Measurement of Total Cell Recovery Is Crucial in the Evaluation of New Macromolecular Cryoprotectants. Biomacromolecules 2020, 21, 2864–2873. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | B1 | B2 | SP1 | SP2 | SP3 | SP4 | SP5 | SP6 | TFDF1 | TFDF2 |
| Cell line | C59 | C113 | C59 | C59 | C59 | C113 | C113 | C113 | C59 | C113 |
| VCCmax [106 cells/mL] | 4.9 | 3.1 | 14.0 | 29.5 | 41.7 | 12.4 | 16.6 | 13.3 | 20.1 | 8.2 |
| HAmax [log10(HAU/100 µL)] | 3.0 | 3.3 | 3.1 | 3.5 | 3.4 | 2.87 | 3.3 | 3.6 | 3.7 | 3.5 |
| HAacc [log10(HAU/100 µL)] | 3.0 | 3.3 | 3.4 | 3.7 | 3.9 | 3.2 | 3.6 | 3.8 | 4.1 | 4.1 |
| TCID50,max [108 TCID50/mL] | 10.0 | 13.0 | 17.8 | 42.2 | 75.0 | 0.1 | 3.2 | 17.8 | 78.1 | 27.7 |
| vir totHA [1011 vp] | 106 | 250 | 16 | 18 | 20 | 8 | 16 | 28 | 3000 | 3060 |
| CSVYHA [vp/cell] | 3900 | 14,680 | 1755 | 1949 | 1166 | 2218 | 3148 | 7281 | 11,506 | 28,882 |
| wv [mL] | 550 | 550 | 30 | 30 | 30 | 30 | 30 | 30 | 1300 | 1300 |
| STYHA [1012 vp/L/d] | 3.2 | 7.6 | 11.1 | 11.1 | 10.0 | 3.1 | 5.2 | 9.3 | 32.7 | 30.5 |
| VVPHA [1012 vp/L/d] | 3.2 | 7.6 | 3.3 | 1.4 | 0.7 | 0.6 | 0.5 | 0.7 | 3.7 | 2.3 |
| Experiment | Control | IST1 | IST2 | IST3 | IST4 | IST5 | IST6 | IFB1 | IFB2 | IFB3 | IFB4 |
| VCC N-1/Inoc. [106 cells/mL] | 10 | 20 | 30 | 40 | 40 O2 lim. | 60 | 10 | 20 | 10 O2 lim. | 20 O2 lim. | |
| VCCmax [106 cells/mL] | 4.5 ±0.1 | 4.3 ±0.2 | 5.9 ±0.1 | 5.7 ±0.1 | 4.8 ±0.1 | 4.5 ±0.1 | 5.1 ±0.2 | 15.3 ±0.2 | 25.7 ±0.2 | 11.4 ±0.4 | 21.4 ±0.4 |
| HAmax [log10(HAU/100 µL)] | 2.8 ±0.1 | 2.8 ±0.1 | 2.9 ±0.1 | 2.7 ±0.1 | 2.8 ±0.1 | 2.6 ±0.1 | 2.7 ±0.1 | 2.9 ±0.1 | 3.0 ±0.3 | 2.9 ±0.1 | n.d. |
| TCID50,max [108 TCID50/mL] | 3.5 ±2.0 | 4.7 ±0.8 | 5.0 ±2.3 | 3.3 ±2.1 | 2.1 ±1.2 | 0.2 ±0.1 | 2.7 ±1.5 | 5.3 ±2.0 | 1.0 ±0.3 | 2.6 ±0.5 | <0.1 |
| vir totHA [1011 vp] | 4.1 ±0.4 | 3.6 ±0.2 | 4.8 ±0.5 | 3.3 ±0.4 | 3.8 ±0.9 | 1.4 ±0.1 | 3.3 ±0.2 | 4.7 ±0.7 | 7.3 ±1.2 | 5.4 ±0.7 | n.d. |
| CSVYHA [vp/cell] | 3024 ±393 | 2805 ±209 | 2740 ±296 | 1896 ±261 | 2287 ±519 | 1015 ±77 | 1972 ±106 | 1009 ±173 | 743 ±373 | 1575 ±238 | n.d. |
| wv [mL] | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 +7 | 30 +13.5 | 30 +7 | 30 +13.2 |
| STYHA [1012 vp/L/d] | 2.8 ±0.3 | 2.6 ±0.1 | 3.6 ±0.4 | 2.3 ±0.3 | 2.6 ±0.6 | 0.7 ±0.1 | 2.2 ±0.1 | 5.2 ±0.8 | 8.2 ±2.8 | 6.0 ±0.8 | n.d. |
| VVPHA [1012 vp/L/d] | 2.8 ±0.3 | 2.6 ±0.1 | 3.6 ±0.4 | 2.3 ±0.3 | 2.6 ±0.6 | 0.7 ±0.1 | 2.2 ±0.1 | 4.3 ±0.7 | 5.7 ±2.7 | 4.8 ±0.6 | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinnecker, T.; Wicke, E.; Reichl, U.; Göbel, S.; Genzel, Y. Seed Train Intensification and TFDF-Based Perfusion for MDCK Cell-Based Influenza A Virus Production. Processes 2025, 13, 1286. https://doi.org/10.3390/pr13051286
Zinnecker T, Wicke E, Reichl U, Göbel S, Genzel Y. Seed Train Intensification and TFDF-Based Perfusion for MDCK Cell-Based Influenza A Virus Production. Processes. 2025; 13(5):1286. https://doi.org/10.3390/pr13051286
Chicago/Turabian StyleZinnecker, Tilia, Emelie Wicke, Udo Reichl, Sven Göbel, and Yvonne Genzel. 2025. "Seed Train Intensification and TFDF-Based Perfusion for MDCK Cell-Based Influenza A Virus Production" Processes 13, no. 5: 1286. https://doi.org/10.3390/pr13051286
APA StyleZinnecker, T., Wicke, E., Reichl, U., Göbel, S., & Genzel, Y. (2025). Seed Train Intensification and TFDF-Based Perfusion for MDCK Cell-Based Influenza A Virus Production. Processes, 13(5), 1286. https://doi.org/10.3390/pr13051286