
The Effects of Solvent and Added Bases on the Protection of Benzylamines with Carbon Dioxide

Abstract
:1. Introduction
2. Experimental Section
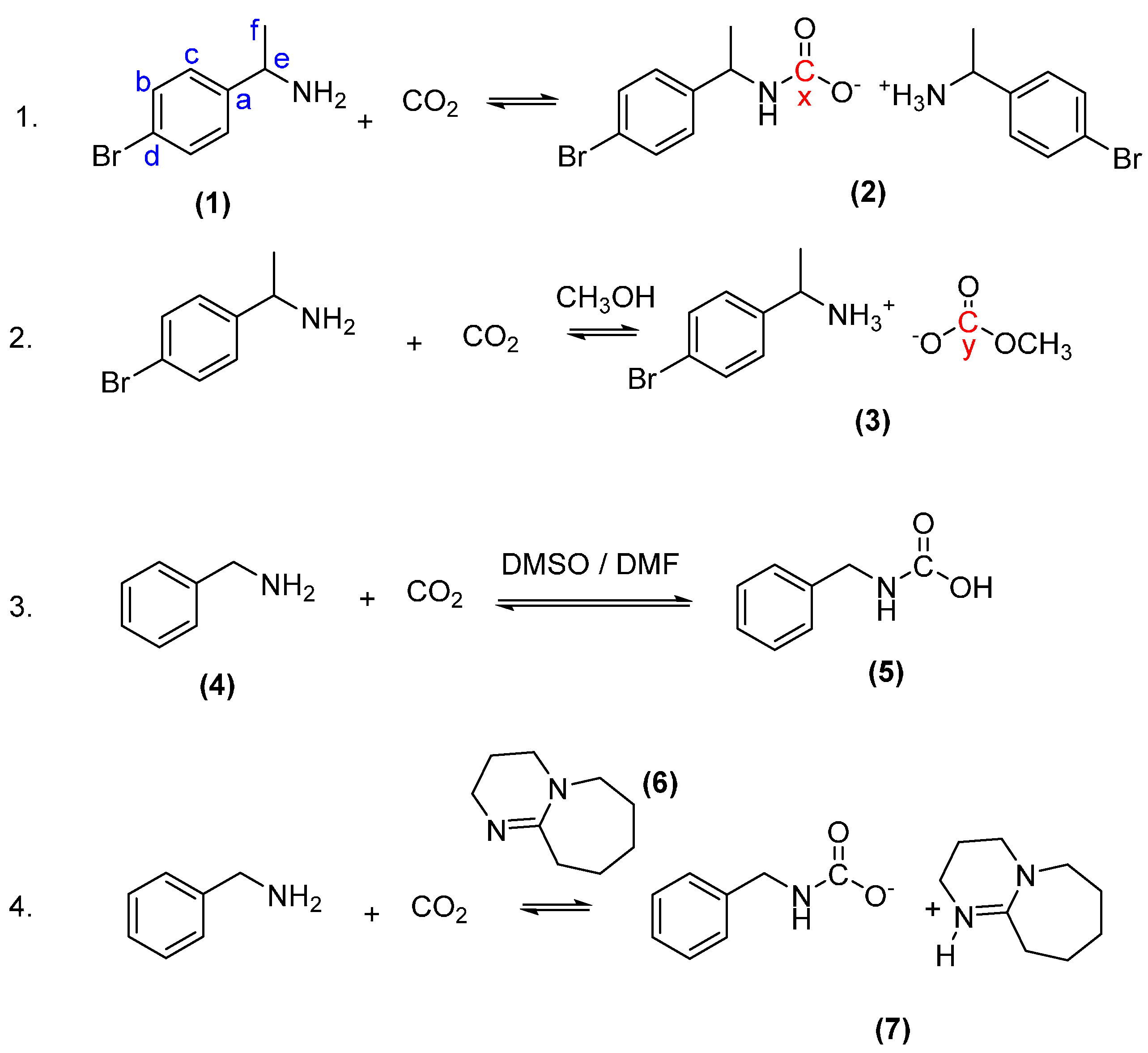
2.1. Reactions with 1-(4-Bromophenyl)ethan-1-Amine (1) and Carbon Dioxide to Form 1-(4-Bromophenyl)ethan-1-Aminium (1-(4-Bromophenyl)Ethyl)Carbamate (Ammonium Carbamate Salt (2))
2.2. Reaction with 1-(4-Bromophenyl)ethan-1-Amine and Carbon Dioxide to Form 1-(4-Bromophenyl)ethan-1-Aminium Methyl Carbonate (Ammonium Carbamate Salt (3))
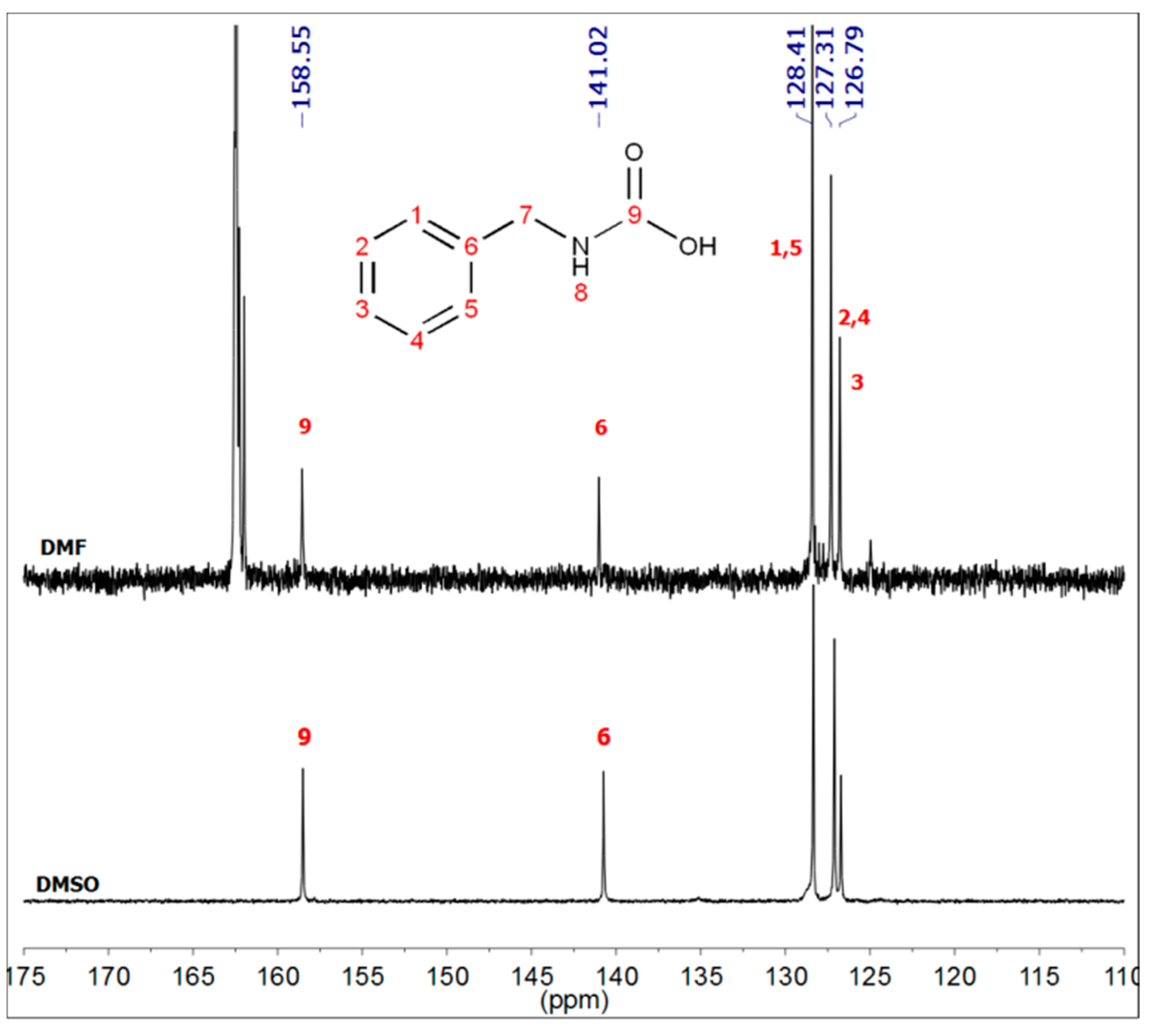
2.3. Synthesis of Benzylcarbamic Acid (5)
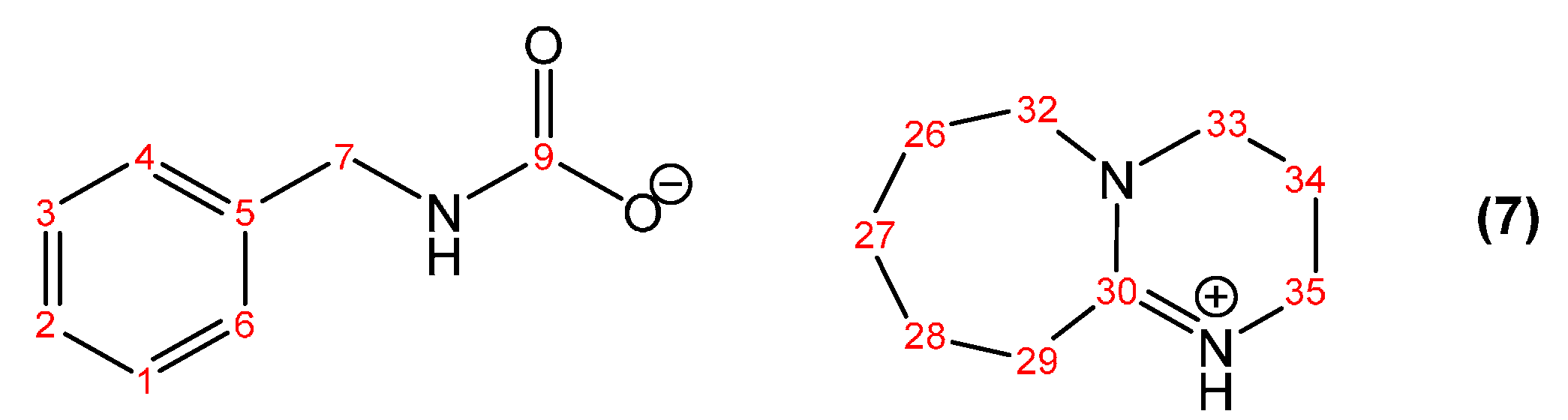
2.4. Synthesis of the DBU Salt of Benzylcarbamic Acid (7)
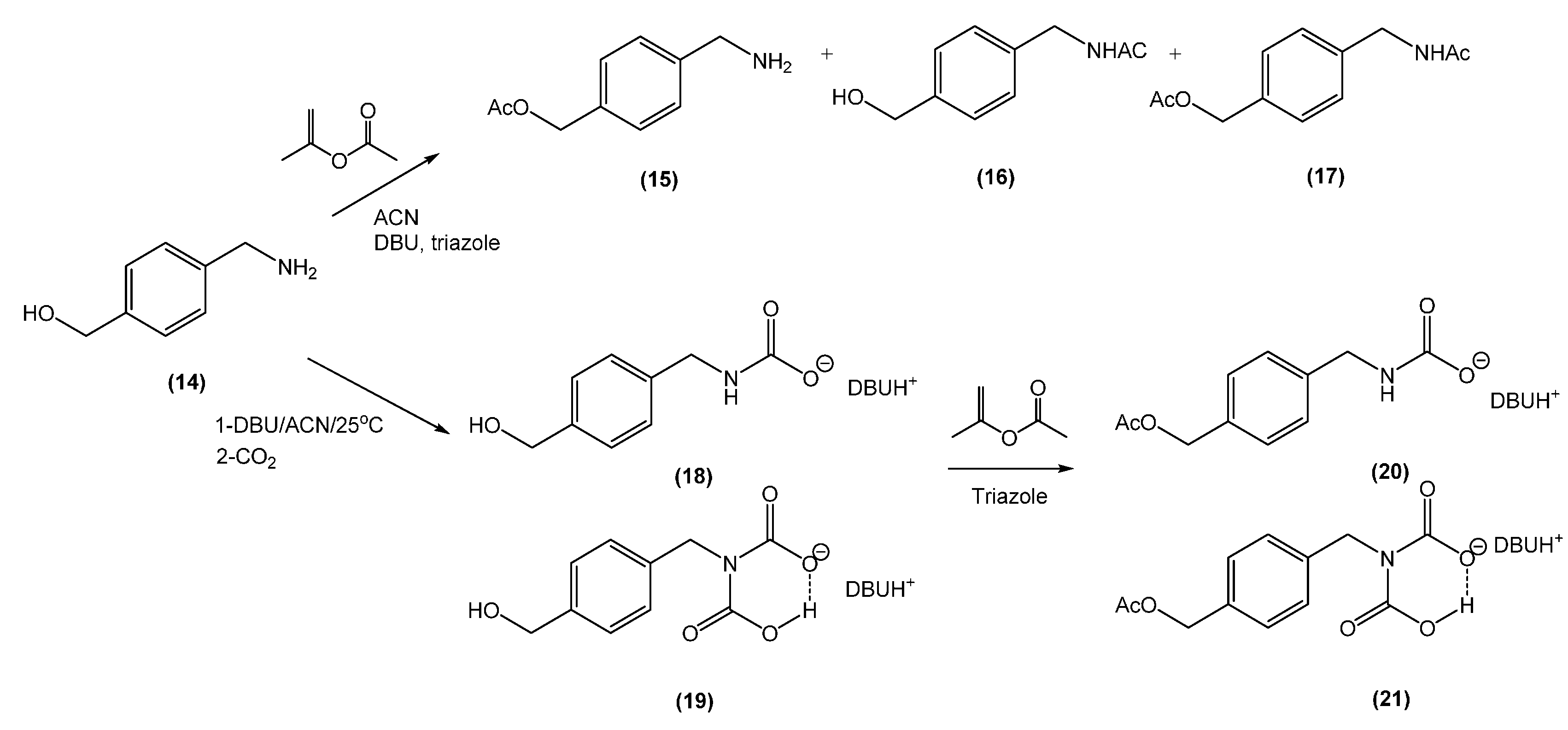
2.5. Synthesis of (4-(Aminomethyl)Phenyl)Methanol (14)
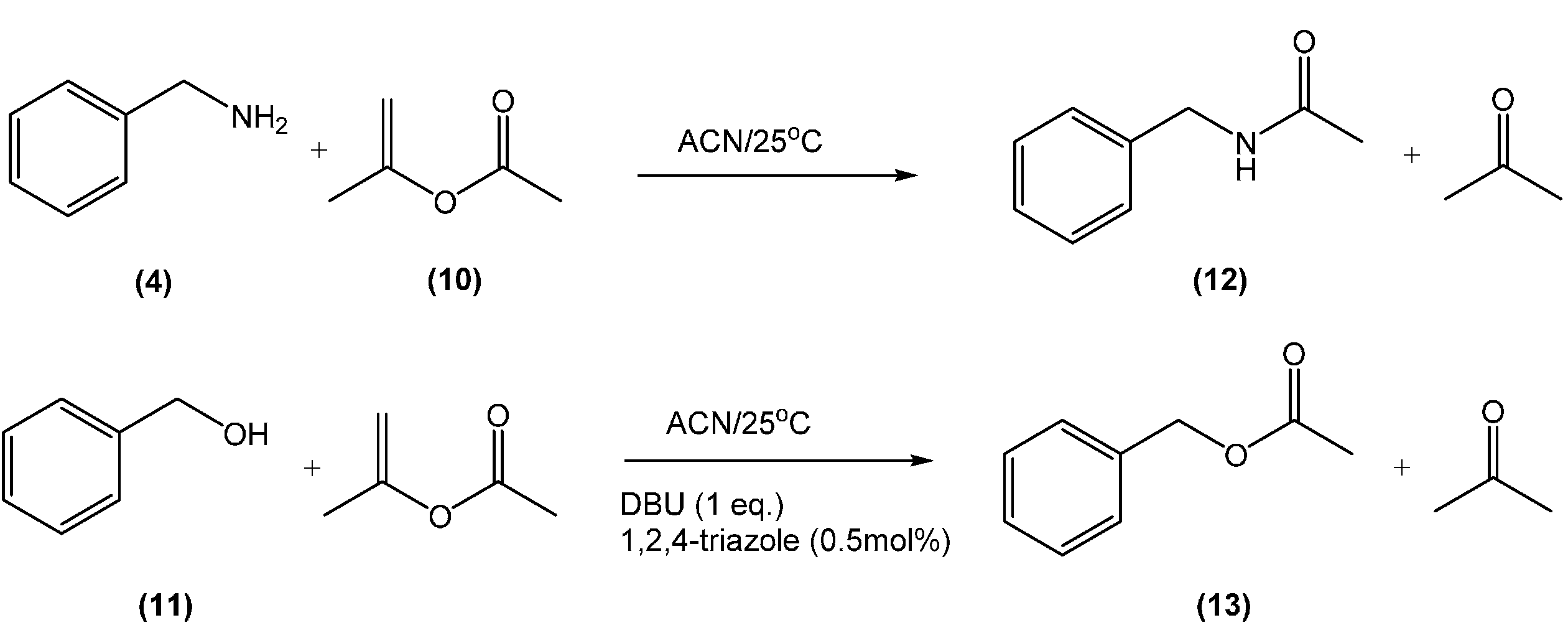
2.6. Procedure for the Acylation Reactions
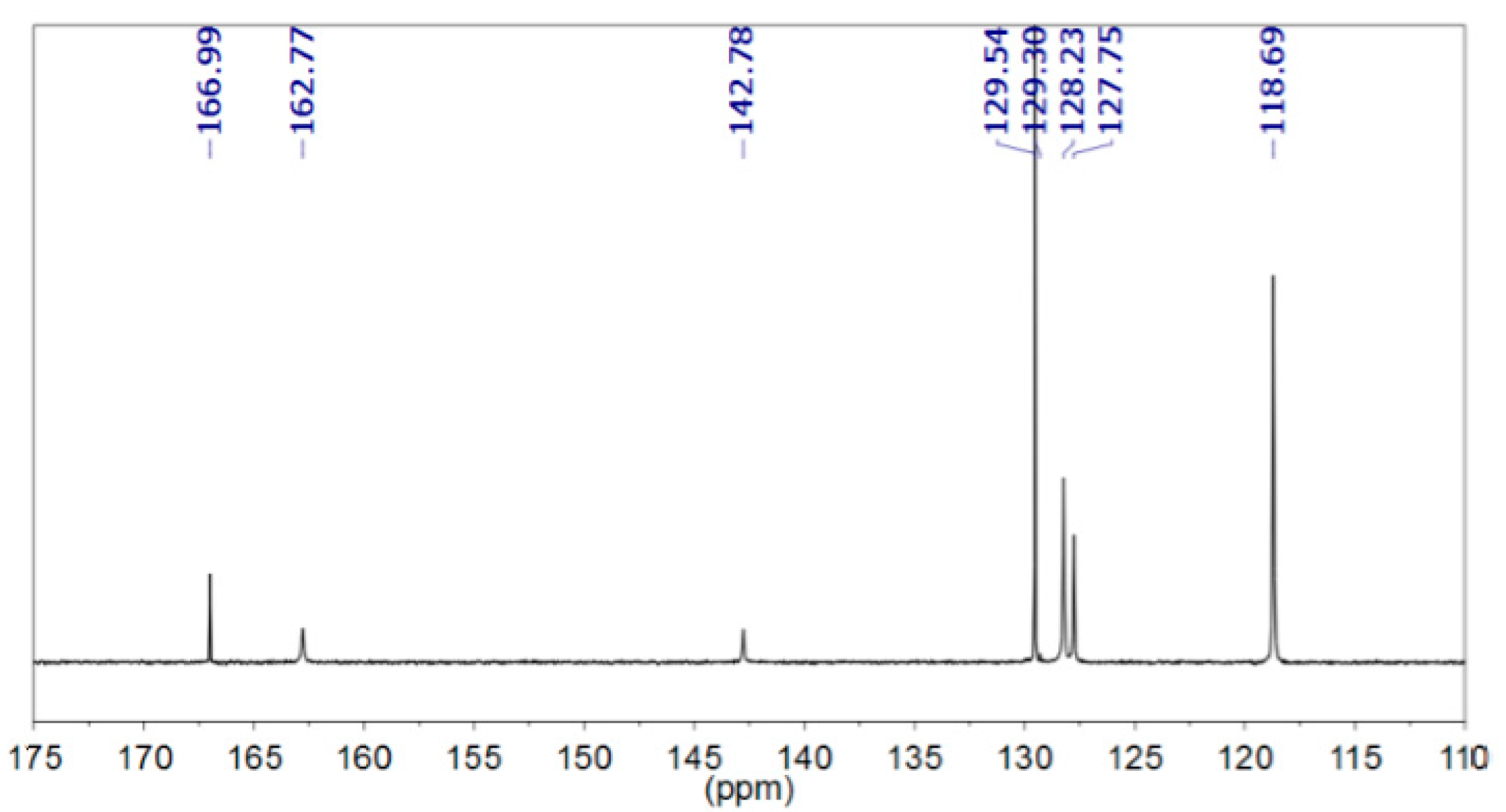
2.7. NMR Analyses
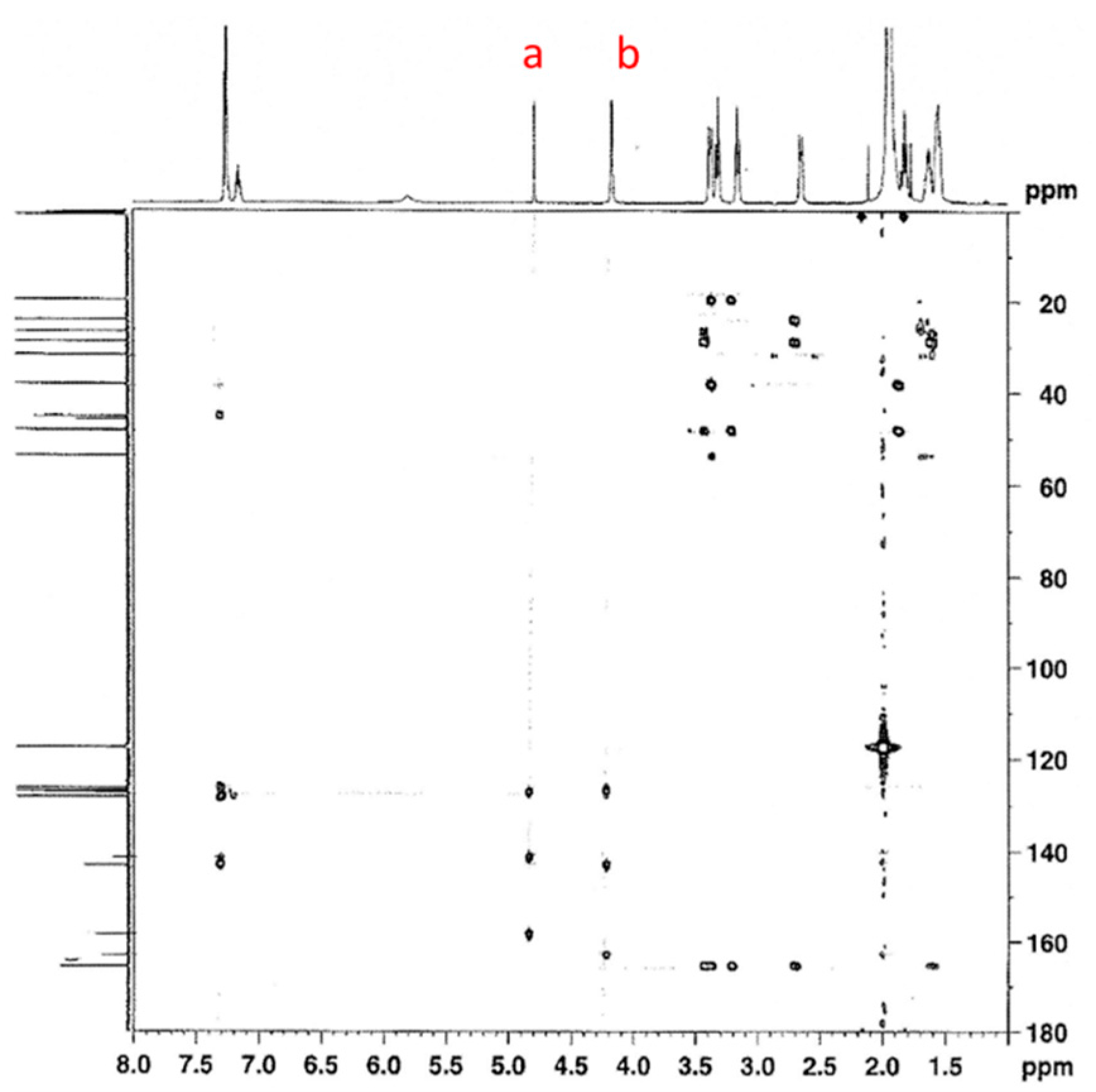
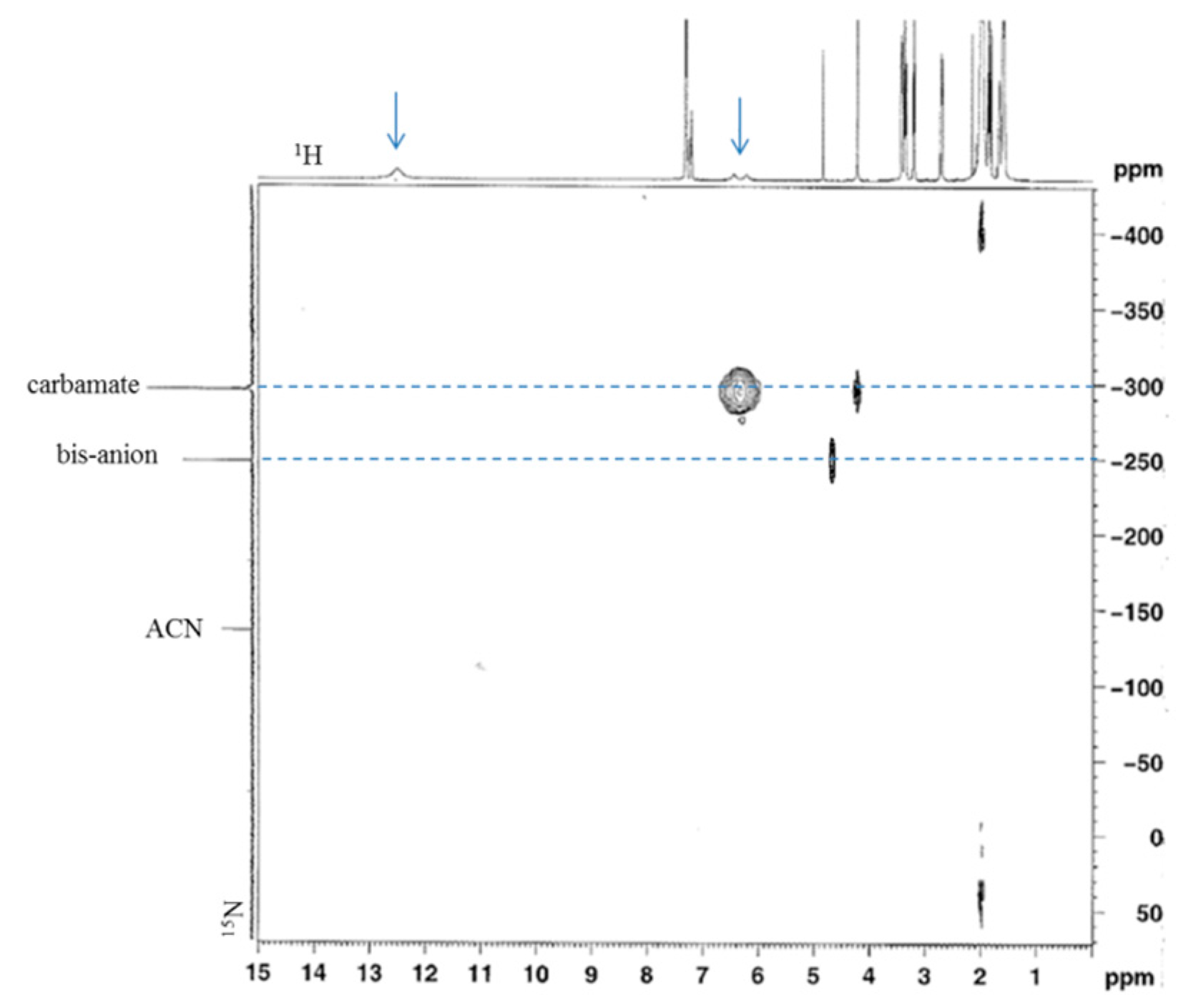
3. Results and Discussion
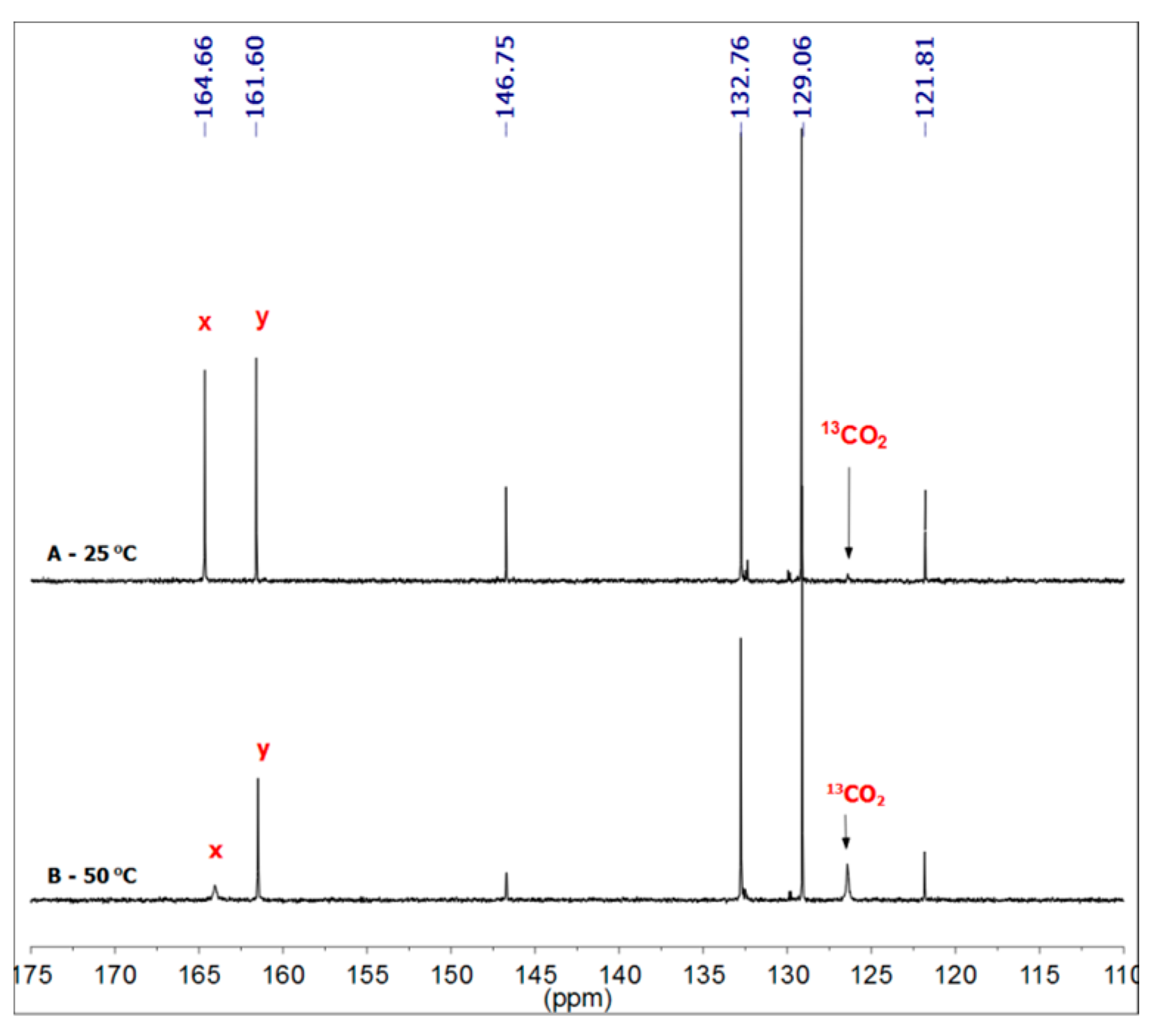
3.1. Reactions of Benzylamines and CO2—the Protection Step




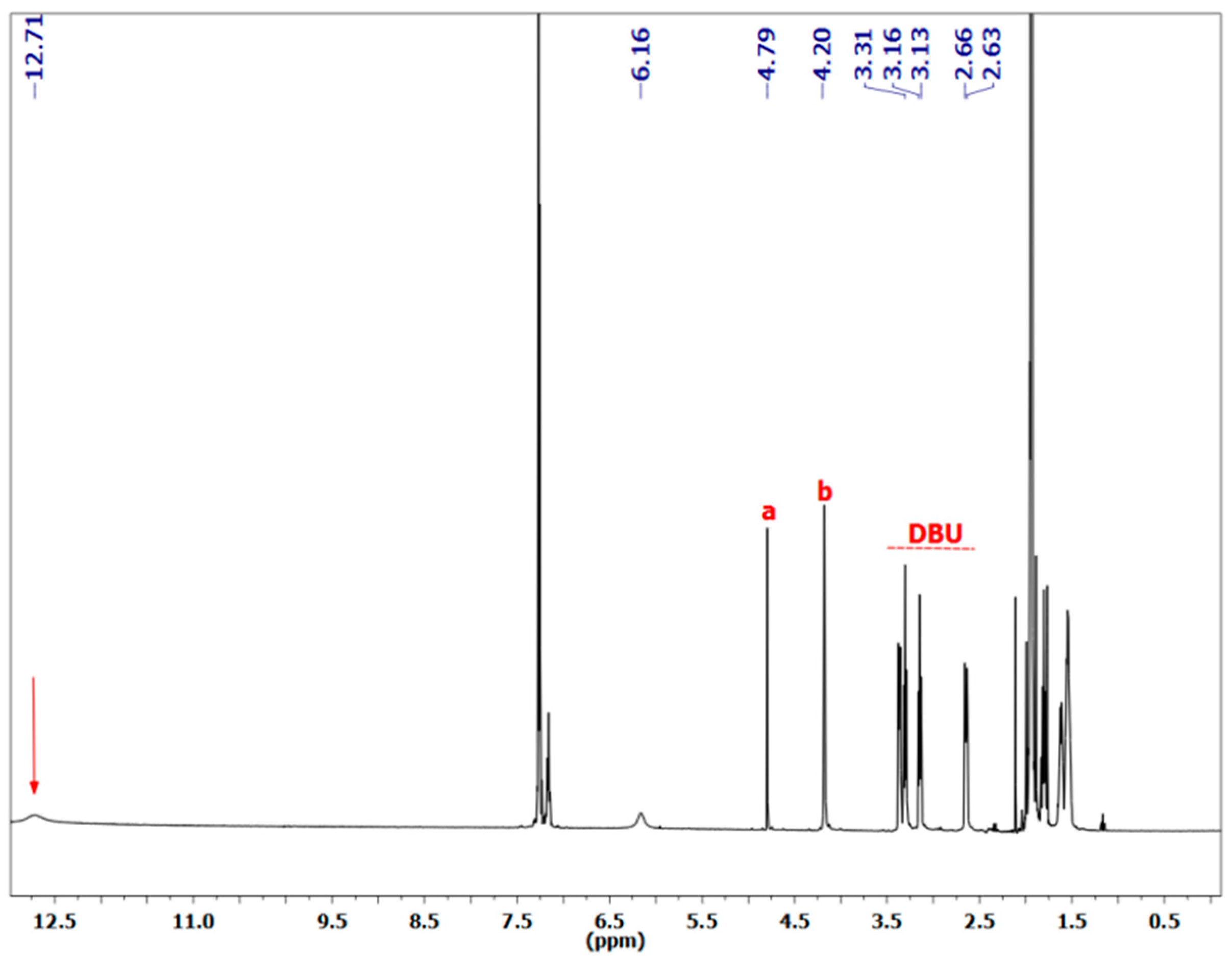
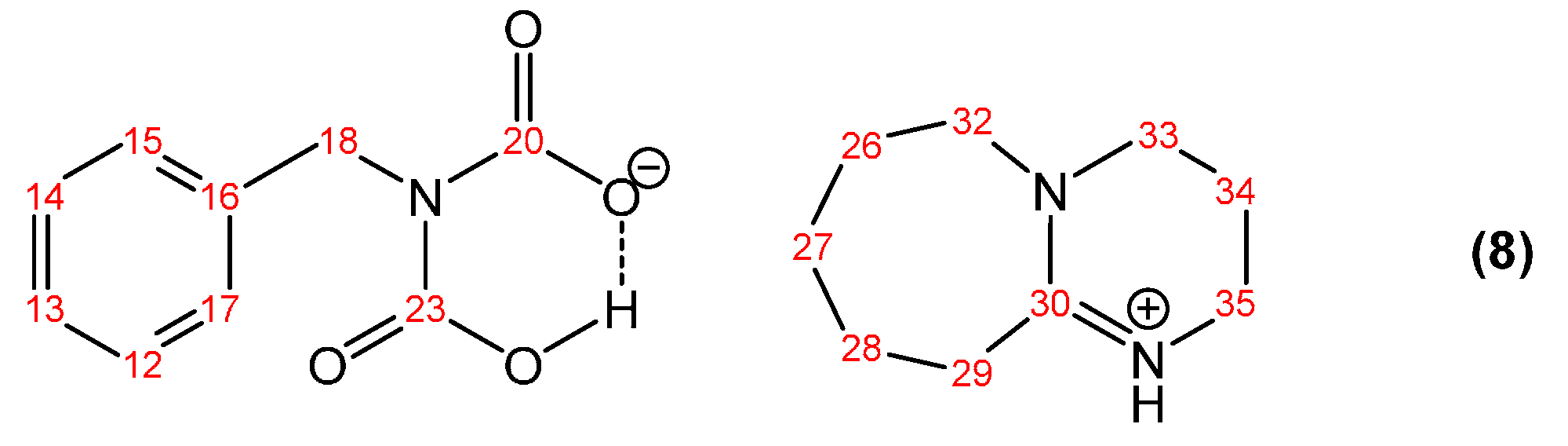
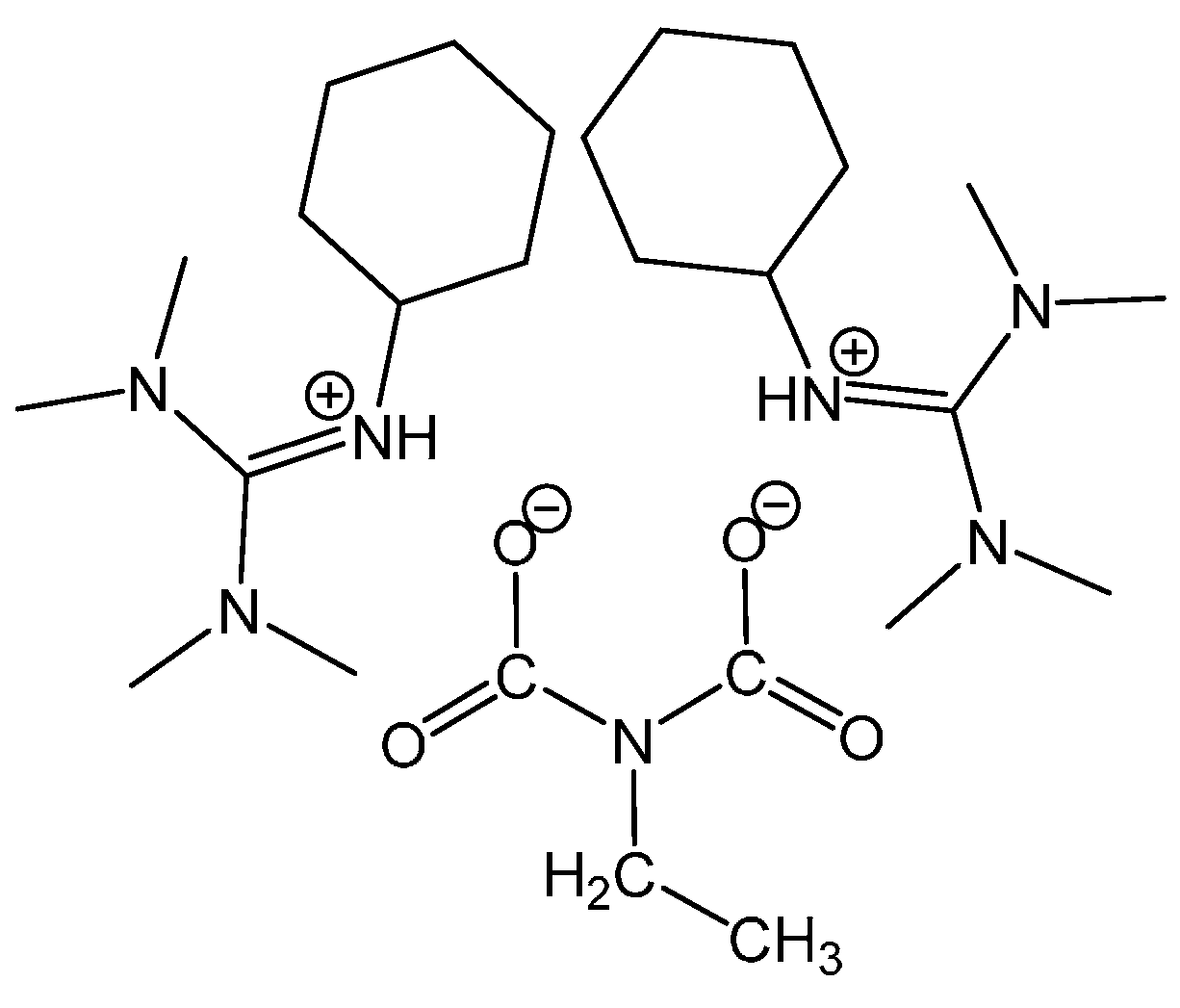
3.1.1. Reaction of Benzylamine with 1 Equivalent of DBU
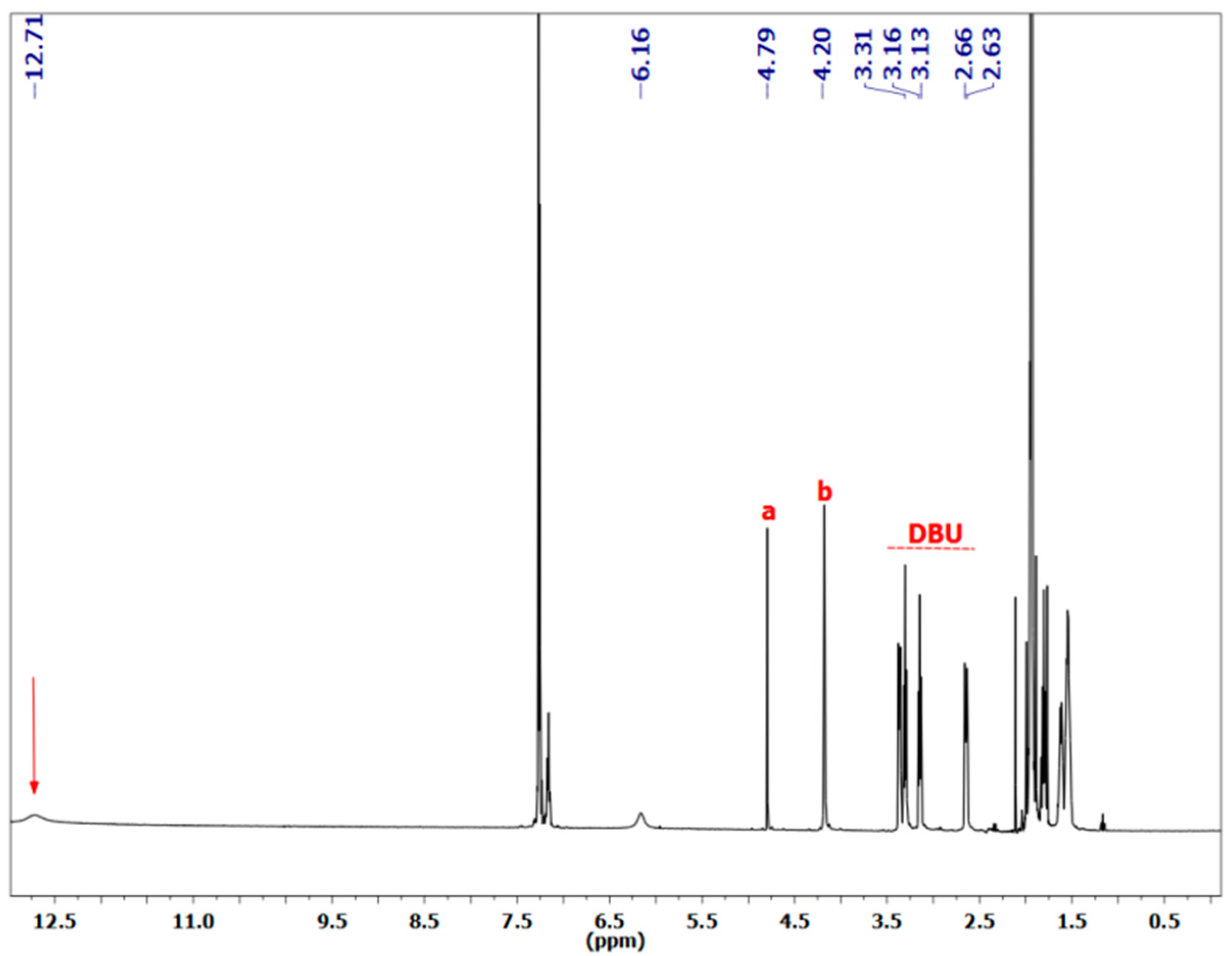


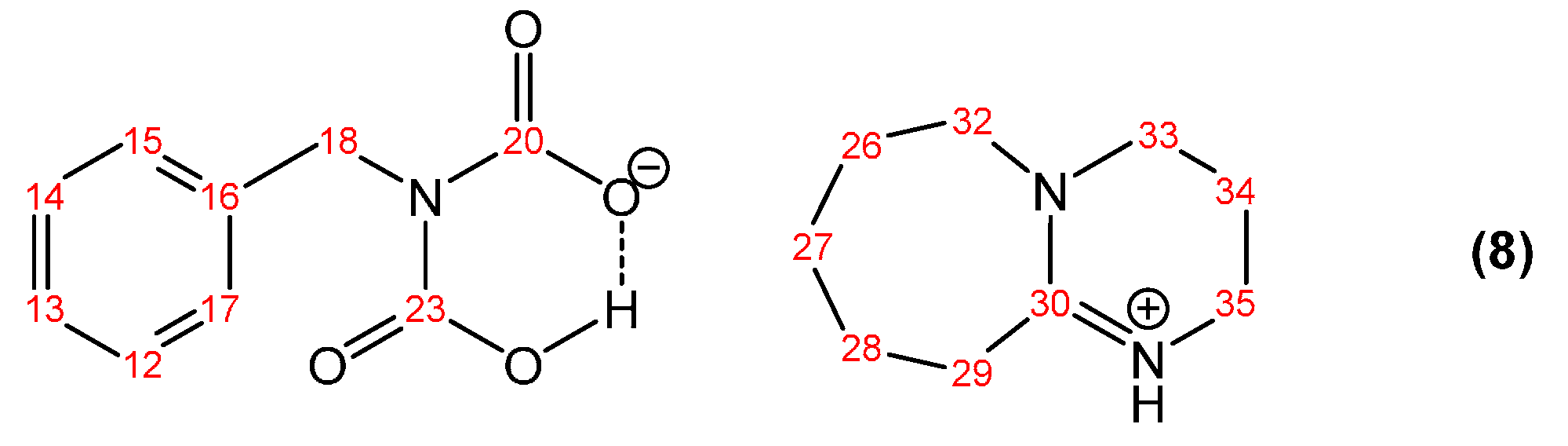


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Assignment | Species | Integration |
|---|---|---|
| 30 | DBUH+ | 1 |
| 9 | Carbamate | 0.54 |
| 20/23 | Bicarbamate | 0.57 |
| 5 | Carbamate | 0.48 |
| 16 | Bicarbamate | 0.23 |
| 7 | Carbamate | 0.49 |
| 18 | Bicarbamate | 0.21 |


3.1.2. Reaction of Benzylamine with 0.5 Equivalent of DBU


3.2. In situ Protection of Amines with CO2


| Entry | CO2 | DBU (eq) | Iso-PA (eq) | Time (h) | O-Product b (%) | N-Product b (%) |
|---|---|---|---|---|---|---|
| 1 | No | 1 | 1 | 6 | 40 | 60 |
| 2 | No | 1.2 | 1.7 | 6 | 70 | 100 |
| 3 | Yes | 1 | 1 | 6 | 15 | 0 |
| 4 | Yes | 1.2 | 1.7 | 6 | 30 | 0 |
| 5 a | Yes | 1.2 | 1.7 | 24 | 80 | 0 |
3.3. Thermal Treatment of the Carbamate-Amidinium Species—the Reversal Step
| Temperature (°C) | Time (h) | Amount of Reversal by NMR Integration (%) |
|---|---|---|
| 60 | 1 | 63 |
| 60 | 2 | 81 |
| 70 | 0.5 | 75 |
| 70 | 1 | 100 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fischer, H.; Gyllenhaal, O.; Vessman, J.; Albert, K. Reaction monitoring of aliphatic amines in supercritical carbon dioxide by proton nuclear magnetic resonance spectroscopy and implications for supercritical fluid chromatography. Anal. Chem. 2003, 75, 622–626. [Google Scholar] [CrossRef]
- Frankel, M.; Katchalski, E. Derivatives of N-carboxy-α-amino acid esters. J. Am. Chem. Soc. 1943, 65, 1670–1674. [Google Scholar] [CrossRef]
- George, M.; Weiss, R.G. Chemically reversible organogels: Aliphatic amines as “latent” gelators with carbon dioxide. J. Am. Chem. Soc. 2001, 123, 10393–10394. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Ito, Y.; Horiguchi, M.; Fujita, H. Studies on the solvent dependence of the carbamic acid formation from ω-(1-naphthyl)alkylamines and carbon dioxide. Tetrahedron 2005, 61, 213–229. [Google Scholar] [CrossRef]
- Mcghee, W.; Riley, D.; Christ, K.; Pan, Y.; Parnas, B. Carbon-dioxide as a phosgene replacement—Synthesis and mechanistic studies of urethanes from amines, CO2, and alkyl chlorides. J. Org. Chem. 1995, 60, 2820–2830. [Google Scholar] [CrossRef]
- Rohan, A.L.; Switzer, J.R.; Flack, K.M.; Hart, R.J.; Sivaswamy, S.; Biddinger, E.J.; Talreja, M.; Verma, M.; Faltermeier, S.; Nielsen, P.T.; et al. The synthesis and the chemical and physical properties of non-aqueous silylamine solvents for carbon dioxide capture. Chemsuschem 2012, 5, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Switzer, J.R.; Ethier, A.L.; Flack, K.M.; Biddinger, E.J.; Gelbaum, L.; Pollet, P.; Eckert, C.A.; Liotta, C.L. Reversible ionic liquid stabilized carbamic acids: A pathway toward enhanced CO2 capture. Ind. Eng. Chem. Res. 2013, 52, 13159–13163. [Google Scholar] [CrossRef]
- Switzer, J.R.; Ethier, A.L.; Hart, E.C.; Flack, K.M.; Rumple, A.C.; Donaldson, J.C.; Bembry, A.T.; Scott, O.M.; Biddinger, E.J.; Talreja, M.; et al. Design, synthesis, and evaluation of nonaqueous silylamines for efficient CO2 capture. Chemsuschem 2014, 7, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.B.; Moore, M.B. Reactions of aralkyl amines with carbon dioxide. J. Am. Chem. Soc. 1948, 70, 3865–3866. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Lukac, P.J.; Yu, T.; Weiss, R.G. Reversible, room-temperature, chiral ionic liquids. Amidinium carbamates derived from amidines and amino-acid esters with carbon dioxide. Chem. Mater. 2007, 19, 4761–4768. [Google Scholar] [CrossRef]
- Mohammed, F.S.; Wuttigul, S.; Kitchens, C.L. Dynamic surface properties of amino-terminated self-assembled monolayers incorporating reversible CO2 chemistry. Ind. Eng. Chem. Res. 2011, 50, 8034–8041. [Google Scholar] [CrossRef]
- Kuchurov, I.V.; Vasilʼev, A.A.; Zlotin, S.G. The suzuki-miyaura cross-coupling of bromo- and chloroarenes with arylboronic acids in supercritical carbon dioxide. Mendeleevsk. Commun. 2010, 20, 140–142. [Google Scholar] [CrossRef]
- Shezad, N.; Oakes, R.S.; Clifford, A.A.; Rayner, C.M. Use of fluorinated palladium sources for efficient Pd-catalysed coupling reactions in supercritical carbon dioxide. Tetrahedron Lett. 1999, 40, 2221–2224. [Google Scholar] [CrossRef]
- Shezad, N.; Clifford, A.A.; Rayner, C.T. Pd-catalysed coupling reactions in supercritical carbon dioxide and under solventless conditions. Green Chem. 2002, 4, 64–67. [Google Scholar] [CrossRef]
- Gordon, R.S.; Holmes, A.B. Palladium-mediated cross-coupling reactions with supported reagents in supercritical carbon dioxide. Chem. Commun. 2002. [Google Scholar] [CrossRef]
- Prajapati, D.; Gohain, M. Recent advances in the application of supercritical fluids for carbon-carbon bond formation in organic synthesis. Tetrahedron 2004, 60, 815–833. [Google Scholar] [CrossRef]
- Carroll, M.A.; Holmes, A.B. Palladium-catalysed carbon-carbon bond formation in supercritical carbon dioxide. Chem. Commun. 1998. [Google Scholar] [CrossRef]
- Peeters, A.; Ameloot, R.; de Vos, D.E. Carbon dioxide as a reversible amine-protecting agent in selective michael additions and acylations. Green Chem. 2013, 15, 1550–1557. [Google Scholar] [CrossRef]
- Dijkstra, Z.J.; Doornbos, A.R.; Weyten, H.; Ernsting, J.M.; Elsevier, C.J.; Keurentjes, J.T.F. Formation of carbamic acid in organic solvents and in supercritical carbon dioxide. J. Supercrit. Fluids 2007, 41, 109–114. [Google Scholar] [CrossRef]
- Colombo, R.; Mingozzi, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Carenini, N.; Perego, P.; Zaffaroni, N.; de Cesare, M.; Castiglioni, V.; et al. Synthesis and biological evaluation (in vitro and in vivo) of cyclic arginine-glycine-aspartate (RGD) peptidomimetic-paclitaxel conjugates targeting lntegrin αVβ3. J. Med. Chem. 2012, 55, 10460–10474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainz, S.; Brinkmann, A.; Leitner, W.; Pfaltz, A. Iridium-catalyzed enantioselective hydrogenation of imines in supercritical carbon dioxide. J. Am. Chem. Soc. 1999, 121, 6421–6429. [Google Scholar] [CrossRef]
- Xie, X.; Liotta, C.L.; Eckert, C.A. CO2-protected amine formation from nitrile and imine hydrogenation in gas-expanded liquids. Ind. Eng. Chem. Res. 2004, 43, 7907–7911. [Google Scholar] [CrossRef]
- Pignataro, L.; Boghi, M.; Civera, M.; Carboni, S.; Piarulli, U.; Gennari, C. Rhodium-catalyzed asymmetric hydrogenation of olefins with phthalaphos, a new class of chiral supramolecular ligands. Chem. Eur. J. 2012, 18, 1383–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The reaction of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) with carbon dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef] [PubMed]
- Mcghee, W.; Riley, D. Replacement of phosgene with carbon-dioxide—Synthesis of alkyl carbonates. J. Org. Chem. 1995, 60, 6205–6207. [Google Scholar] [CrossRef]
- Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Isopropenyl acetate, a remarkable, cheap and acylating agent of amines under solvent- and catalyst-free conditions: A systematic investigation. Green Chem. 2012, 14, 2251–2255. [Google Scholar] [CrossRef]
- Yang, X.; Birman, V.B. Acyl transfer catalysis with 1,2,4-triazole anion. Org. Lett. 2009, 11, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ethier, A.L.; Switzer, J.R.; Rumple, A.C.; Medina-Ramos, W.; Li, Z.; Fisk, J.; Holden, B.; Gelbaum, L.; Pollet, P.; Eckert, C.A.; et al. The Effects of Solvent and Added Bases on the Protection of Benzylamines with Carbon Dioxide. Processes 2015, 3, 497-513. https://doi.org/10.3390/pr3030497
Ethier AL, Switzer JR, Rumple AC, Medina-Ramos W, Li Z, Fisk J, Holden B, Gelbaum L, Pollet P, Eckert CA, et al. The Effects of Solvent and Added Bases on the Protection of Benzylamines with Carbon Dioxide. Processes. 2015; 3(3):497-513. https://doi.org/10.3390/pr3030497
Chicago/Turabian StyleEthier, Amy L., Jackson R. Switzer, Amber C. Rumple, Wilmarie Medina-Ramos, Zhao Li, Jason Fisk, Bruce Holden, Leslie Gelbaum, Pamela Pollet, Charles A. Eckert, and et al. 2015. "The Effects of Solvent and Added Bases on the Protection of Benzylamines with Carbon Dioxide" Processes 3, no. 3: 497-513. https://doi.org/10.3390/pr3030497
APA StyleEthier, A. L., Switzer, J. R., Rumple, A. C., Medina-Ramos, W., Li, Z., Fisk, J., Holden, B., Gelbaum, L., Pollet, P., Eckert, C. A., & Liotta, C. L. (2015). The Effects of Solvent and Added Bases on the Protection of Benzylamines with Carbon Dioxide. Processes, 3(3), 497-513. https://doi.org/10.3390/pr3030497