
Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome

Abstract
:1. Introduction
2. Results
2.1. Metabolic Analysis of Single Species Biofilms
- B. thetaiotaomicron: high fluxes of acetate and CO2 and a low flux of propionate
- F. prausnitzii: high fluxes of butyrate, CO2, formate and lactate
- E. coli: high fluxes of acetate, ethanol and formate and a very low flux of succinate
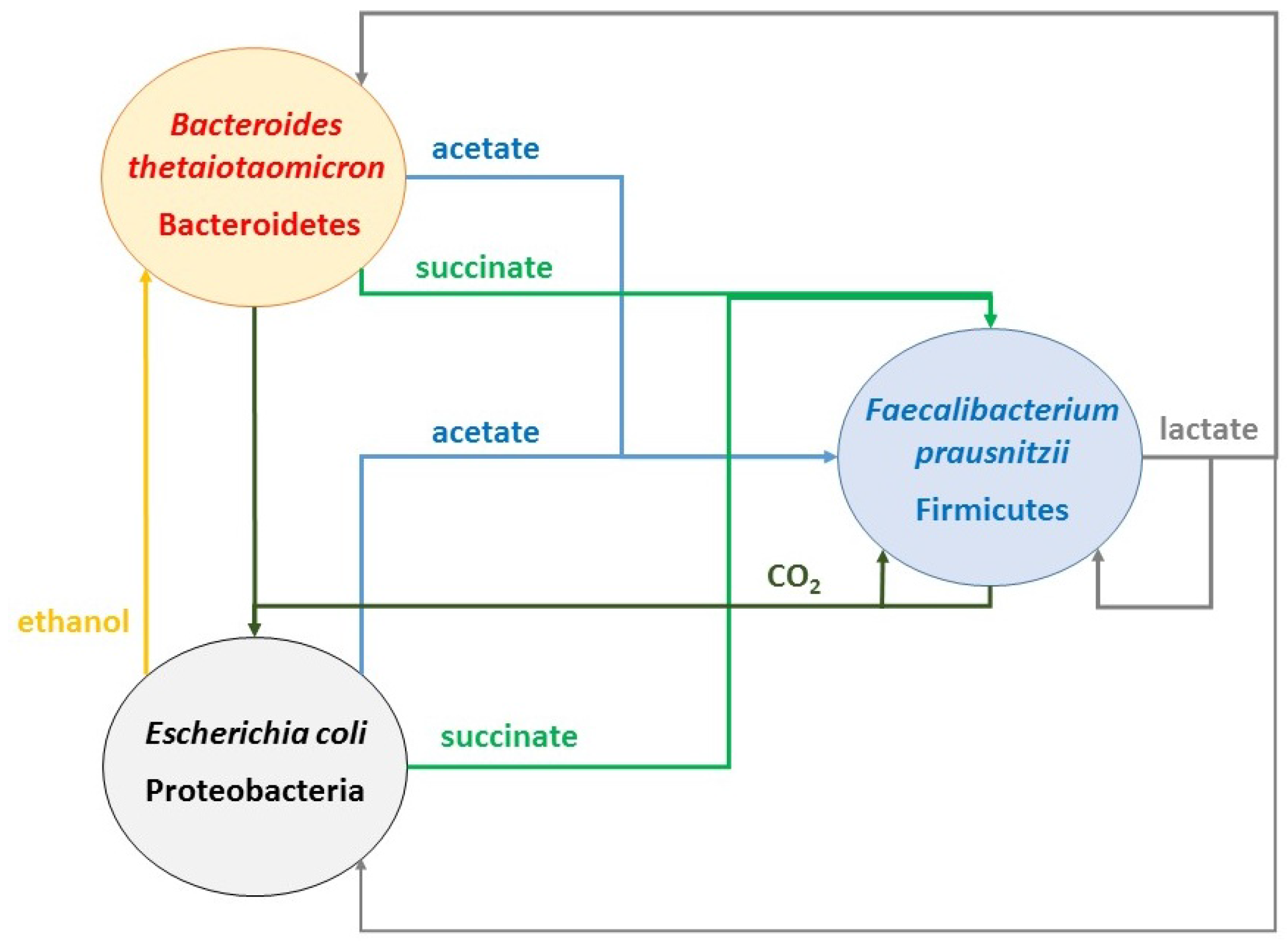
2.2. Predicting Cross-Feeding Relationships in the Multispecies Biofilm
- Acetate: produced by B. thetaiotaomicron and E. coli; consumed by F. prausnitzii
- CO2: produced by F. prausnitzii and B. thetaiotaomicron; consumed by E. coli and F. prausnitzii
- Ethanol: produced by E. coli; consumed by B. thetaiotaomicron
- Formate: produced by E. coli and F. prausnitzii; consumed by F. prausnitzii
- Lactate: produced by F. prausnitzii; consumed by B. thetaiotaomicron and E. coli
- Succinate: produced by B. thetaiotaomicron and E. coli; consumed by E. coli
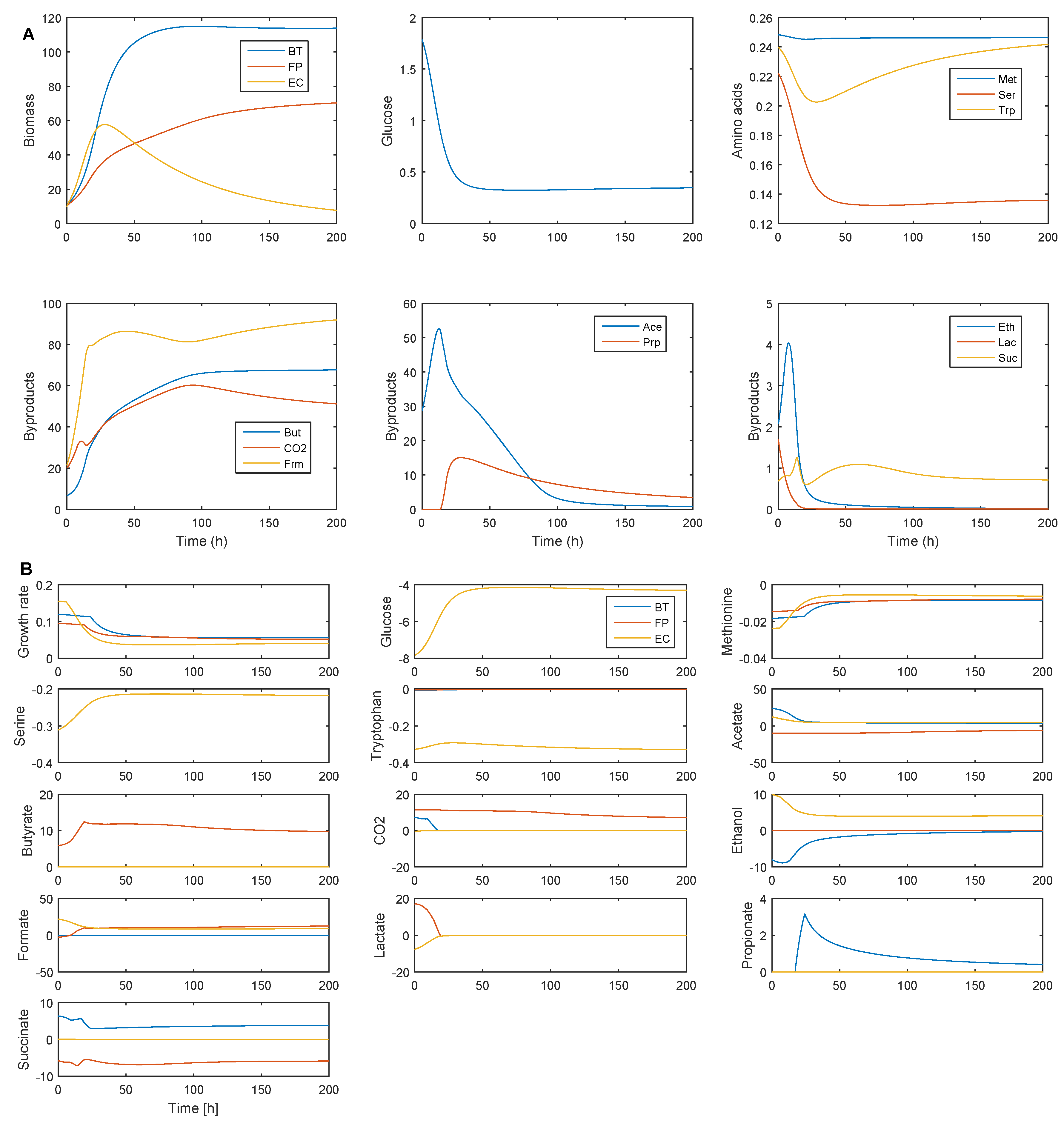
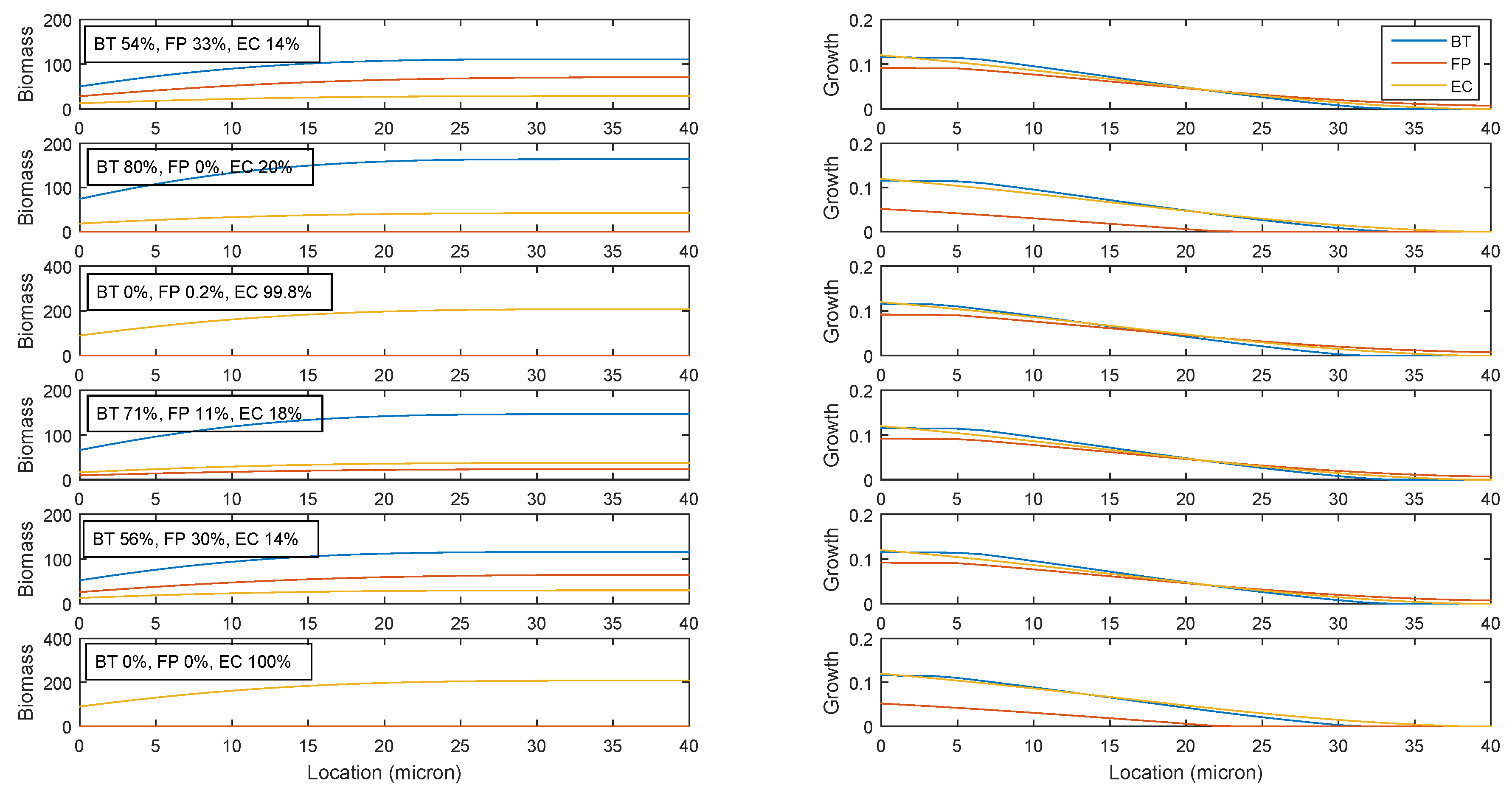
2.3. Establishing Species Coexistence in the Multispecies Biofilm
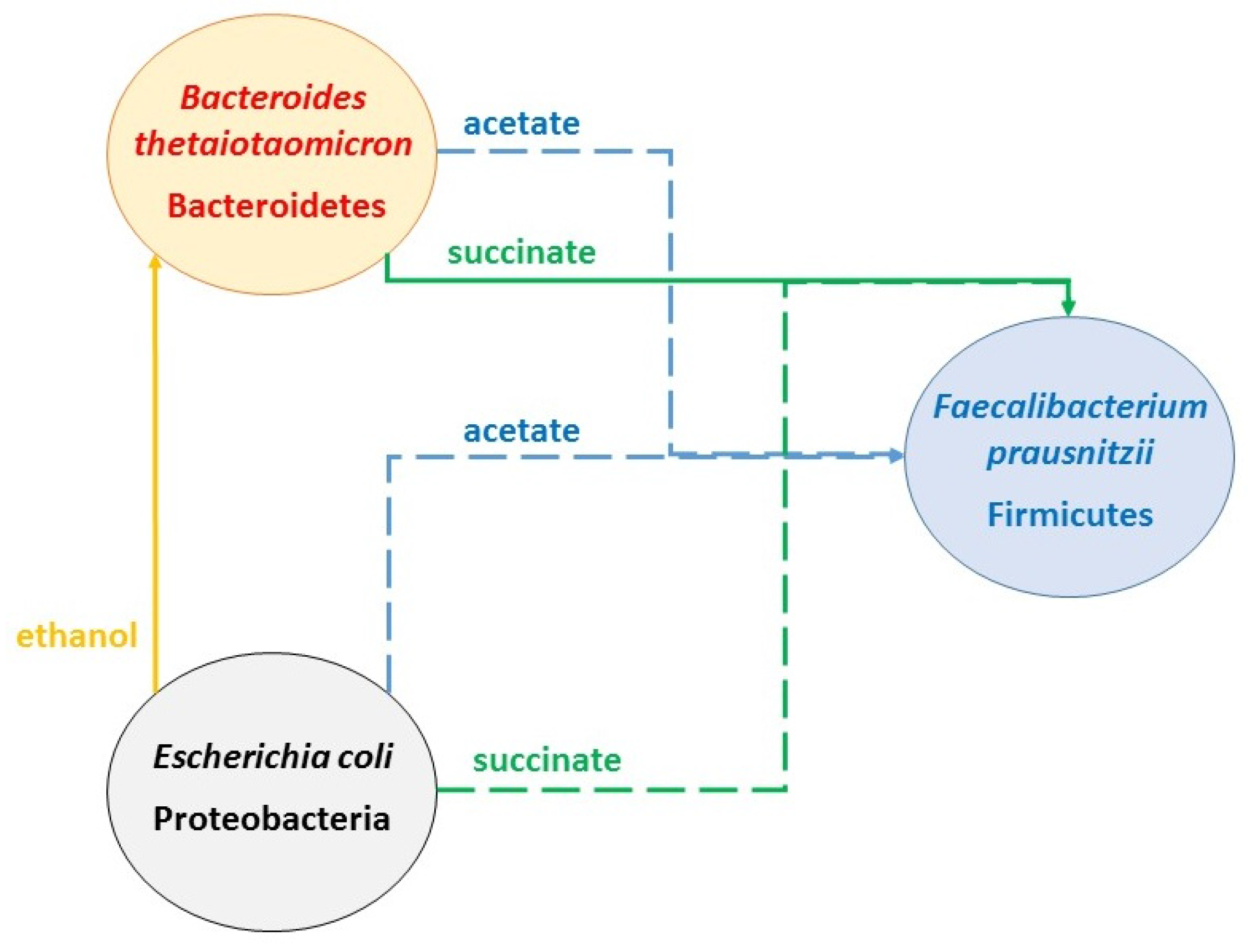
2.4. Identifying Cross-Feeding Relationships Essential for Community Stability
- Nominal case: acetate and succinate produced by B. thetaiotaomicron and E. coli consumed by F. prausnitzii; ethanol produced by E. coli consumed by B. thetaiotaomicron
- Nominal case without acetate consumption by F. prausnitzii
- Nominal case without succinate consumption by F. prausnitzii
- Nominal case without ethanol consumption by B. thetaiotaomicron
- Nominal case without F. prausnitzii consumption of acetate produced by B. thetaiotaomicron
- Nominal case without F. prausnitzii consumption of succinate produced by B. thetaiotaomicron
- Nominal case without F. prausnitzii consumption of acetate produced by E. coli
- Nominal case without F. prausnitzii consumption of succinate produced by E. coli
- No cross feeding
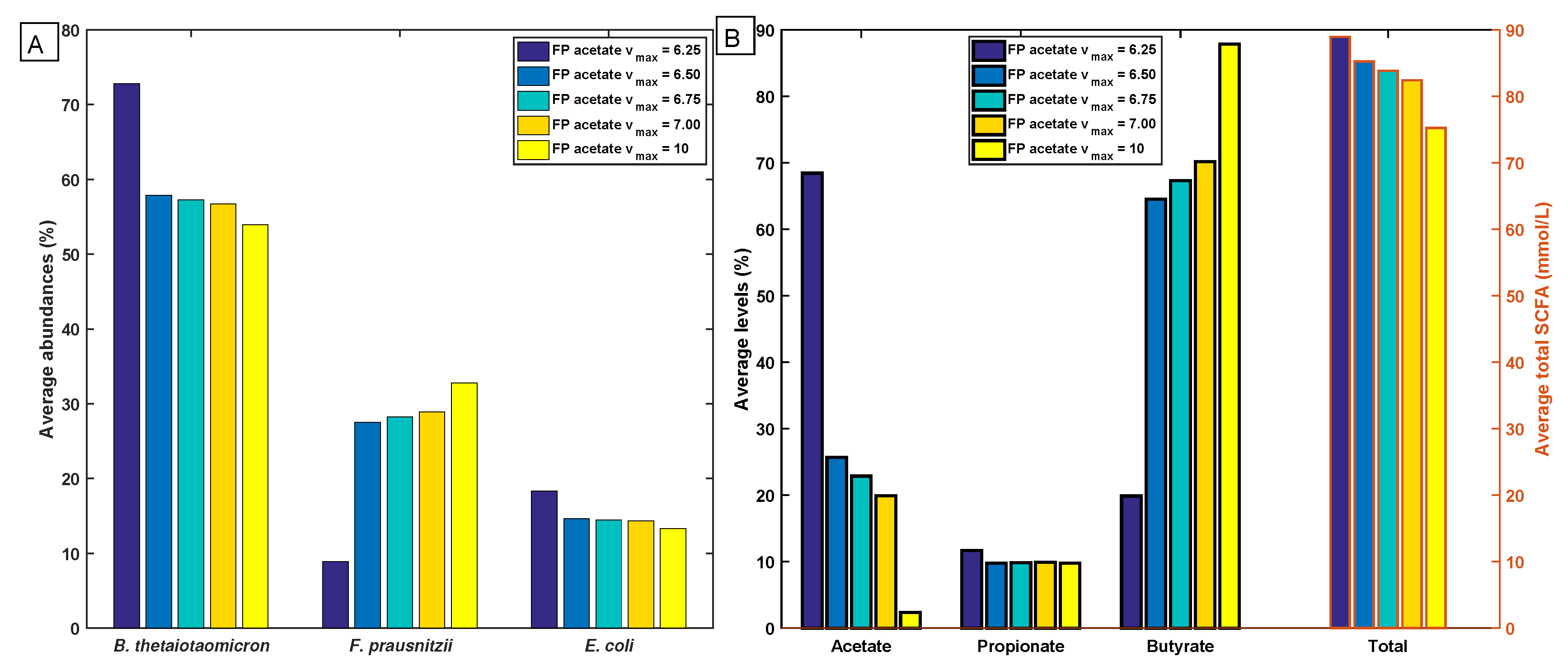
2.5. Robustness of the Cross-Feeding Strategy
2.6. Flexibility of the Cross-Feeding Strategy
3. Discussion
4. Materials and Methods
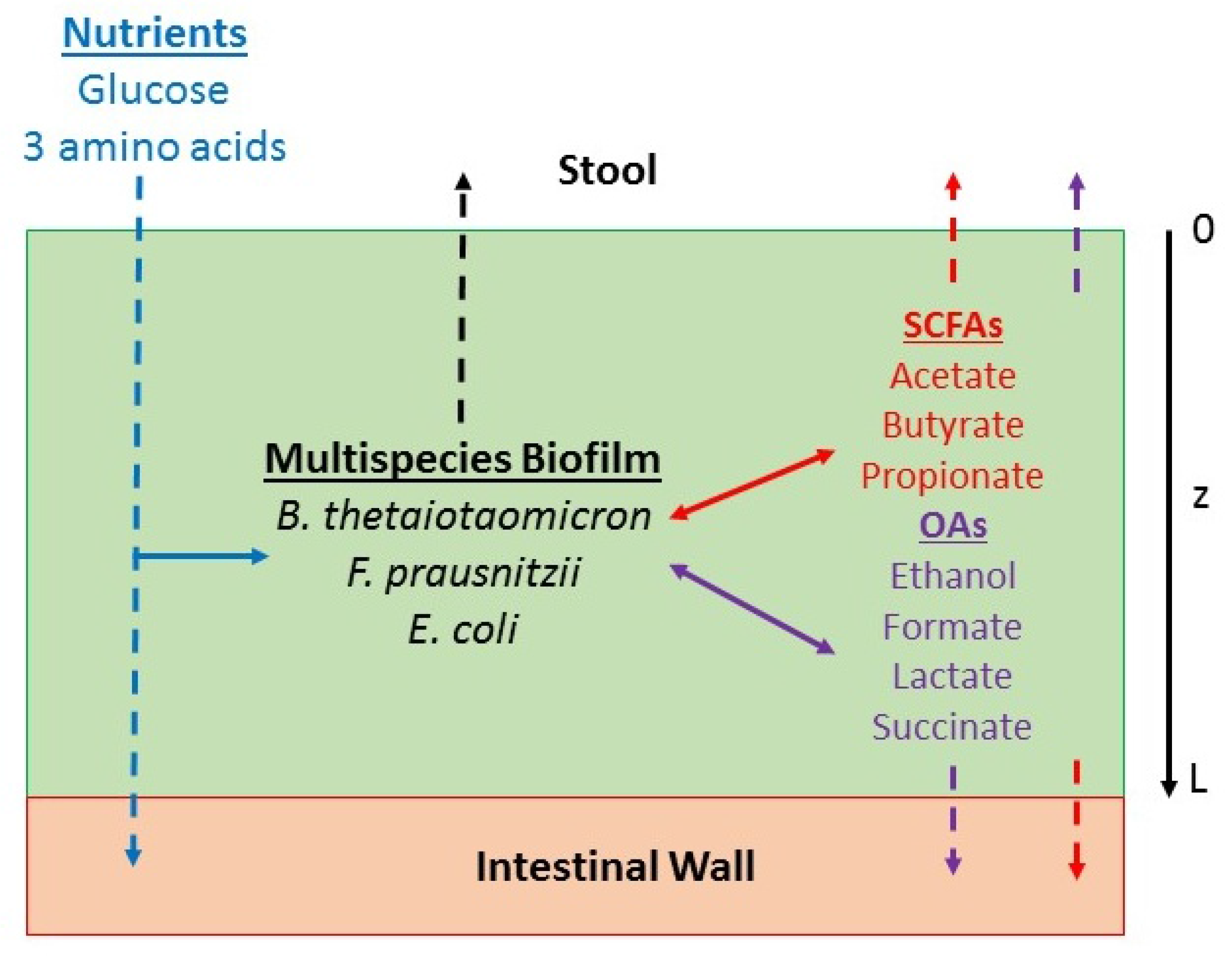
4.1. Multispecies Biofilm Model Formulation
4.2. Model Parameterization and Solution
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | Amino acid |
| Ace | Acetate |
| BT | B. thetaiotaomicron |
| But | Butyrate |
| FBA | Flux balance analysis |
| EC | E. coli |
| Eth | Ethanol |
| FP | F. prausnitzii |
| Frm | Formate |
| Glc | Glucose |
| Lac | Lactate |
| LP | Linear program |
| Met | Methionine |
| OA | Organic acid |
| ODE | Ordinary differential equation |
| PDE | Partial differential equation |
| Prp | Propionate |
| Ser | Serine |
| SFCA | Short chain fatty acid |
| Suc | Succinate |
| Trp | Tryptophan |
References
- Stoodley, P.; Sauer, K.; Davies, D.; Costerton, J.W. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.C.; Carlson, R.P. Microbial consortia engineering for cellular factories: In vitro to in silico systems. Comput. Struct. Biotechnol. J. 2012, 3, e201210017. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 2015, 13, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Hol, F.J.; Galajda, P.; Woolthuis, R.G.; Dekker, C.; Keymer, J.E. The idiosyncrasy of spatial structure in bacterial competition. BMC Res. Notes 2015, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Á.T. Impact of spatial distribution on the development of mutualism in microbes. Front. Microbiol. 2014, 5, 649. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T.; Molin, S. Spatial organization of microbial biofilm communities. Microb. Ecol. 2000, 40, 75–84. [Google Scholar] [PubMed]
- Burmølle, M.; Ren, D.; Bjarnsholt, T.; Sørensen, S.J. Interactions in multispecies biofilms: Do they actually matter? Trends Microbiol. 2014, 22, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, V.; Amar, S.; Segrè, D. Metabolic proximity in the order of colonization of a microbial community. PLoS ONE 2013, 8, e77617. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. A review of experimental measurements of effective diffusive permeabilities and effective diffusion coefficients in biofilms. Biotechnol. Bioeng. 1998, 59, 261–272. [Google Scholar] [CrossRef]
- Woods, J.; Boegli, L.; Kirker, K.R.; Agostinho, A.M.; Durch, A.M.; deLancey Pulcini, E.; Stewart, P.S.; James, G.A. Development and application of a polymicrobial, in vitro, wound biofilm model. J. Appl. Microbiol. 2012, 112, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, T.R.; Bernstein, H.; Lloyd-Randolfi, J.; Jimenez-Taracido, L.; Stewart, P.S.; Carlson, R.P. Robustness analysis of culturing perturbations on Escherichia coli colony biofilm beta-lactam and aminoglycoside antibiotic tolerance. BMC Microbiol. 2010, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef] [PubMed]
- Seth, E.C.; Taga, M.E. Nutrient cross-feeding in the microbial world. Front. Microbiol. 2014, 5, 350. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Merker, H.; Bohl, K.; Reichelt, M.; Schuster, S.; De Figueiredo, L.F.; Kaleta, C.; Kost, C. Fitness and stability of obligate cross-feeding interactions that emerge upon gene loss in bacteria. ISME J. 2014, 8, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Woloszynek, S.; Pastor, S.; Mell, J.; Nandi, N.; Sokhansanj, B.; Rosen, G. Engineering Human Microbiota: Influencing Cellular and Community Dynamics for Therapeutic Applications. Int. Rev. Cell Mol. Biol. 2016, 324, 67–124. [Google Scholar] [PubMed]
- Poltak, S.R.; Cooper, V.S. Ecological succession in long-term experimentally evolved biofilms produces synergistic communities. ISME J. 2011, 5, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Wintermute, E.H.; Silver, P.A. Dynamics in the mixed microbial concourse. Genes Dev. 2010, 24, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Pitts, B.; Lauchnor, E.; Cappitelli, F.; Stewart, P.S. Development of a laboratory model of a phototroph-heterotroph mixed-species biofilm at the stone/air interface. Front. Microbiol. 2015, 6, 1251. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Kaftan, F.; Lang, S.; Svatoš, A.; Germerodt, S.; Kost, C. Privatization of cooperative benefits stabilizes mutualistic cross-feeding interactions in spatially structured environments. ISME J. 2016, 10, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [Green Version]
- Thompson, J.A.; Oliveira, R.A.; Xavier, K.B. Chemical conversations in the gut microbiota. Gut Microbes 2016, 7, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Sonnenburg, J.L. Eating for two: How metabolism establishes interspecies interactions in the gut. Cell Host Microbe 2011, 10, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Gur, T.L.; Bailey, M.T. Effects of stress on commensal microbes and immune system activity. In Microbial Endocrinology: Interkingdom Signaling in Infectious Disease and Health; Springer: New York, NY, USA, 2016; pp. 289–300. [Google Scholar]
- Theriot, C.M.; Young, V.B. Interactions between the gastrointestinal microbiome and Clostridium difficile. Annu. Rew. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- yri, S.F.; Muzes, G.; Sipos, F. Dysbiotic gut microbiome: A key element of Crohn’s disease. Comp. Immunol. Microbiol. Infect. Dis. 2015, 43, 36–49. [Google Scholar] [CrossRef] [PubMed]
- John, G.K.; Mullin, G.E. The gut microbiome and obesity. Curr. Oncol. Rep. 2016, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Bäckhed, F. The gut microbiota and metabolic disease: Current understanding and future perspectives. J. Intern. Med. 2016, 280, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Koopen, A.M.; Groen, A.K.; Nieuwdorp, M. Human microbiome as therapeutic intervention target to reduce cardiovascular disease risk. Curr. Opin. Lipidol. 2016, 27, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.T.; Asquith, M.J. The microbiome: A revolution in treatment for rheumatic diseases? Curr. Rheumatol. Rep. 2016, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Manzat-Saplacan, R.M.; Mircea, P.A.; Balacescu, L.; Chira, R.I.; Berindan-Neagoe, I.; Balacescu, O. Can we change our microbiome to prevent colorectal cancer development? Acta Oncol. 2015, 54, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Clarke, G.; Berk, M.; Jacka, F.N. The gut microbiome and diet in psychiatry: Focus on depression. Curr. Opin. Psychiatry 2015, 28, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Chambers, E.; Morrison, D.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Schloissnig, S.; Arumugam, M.; Sunagawa, S.; Mitreva, M.; Tap, J.; Zhu, A.; Waller, A.; Mende, D.R.; Kultima, J.R.; Martin, J.; et al. Genomic variation landscape of the human gut microbiome. Nature 2013, 493, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69. [Google Scholar] [CrossRef] [PubMed]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Stares, M.D.; Connor, T.R.; Raisen, C.; Goulding, D.; Rad, R.; Schreiber, F.; Brandt, C.; et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012, 8, e1002995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seekatz, A.M.; Young, V.B. Clostridium difficile and the microbiota. J. Clin. Investig. 2014, 124, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Estrela, S.; Brown, S.P. Metabolic and demographic feedbacks shape the emergent spatial structure and function of microbial communities. PLoS Comput. Biol. 2013, 9, e1003398. [Google Scholar] [CrossRef] [PubMed]
- Brenner, K.; You, L.; Arnold, F.H. Engineering microbial consortia: A new frontier in synthetic biology. Trends Biotechnol. 2008, 26, 483–489. [Google Scholar] [CrossRef] [PubMed]
- De Roy, K.; Marzorati, M.; Van den Abbeele, P.; Van de Wiele, T.; Boon, N. Synthetic microbial ecosystems: An exciting tool to understand and apply microbial communities. Environ. Microbiol. 2014, 16, 1472–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shong, J.; Diaz, M.R.J.; Collins, C.H. Towards synthetic microbial consortia for bioprocessing. Curr. Opin. Biotechnol. 2012, 23, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Jagmann, N.; Philipp, B. Reprint of design of synthetic microbial communities for biotechnological production processes. J. Biotechnol. 2014, 192, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Boedicker, J.Q.; Choi, J.W.; Ismagilov, R.F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc. Natl. Acad. Sci. USA 2008, 105, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Hoek, T.A.; Axelrod, K.; Biancalani, T.; Yurtsev, E.A.; Liu, J.; Gore, J. Resource availability modulates the cooperative and competitive nature of a microbial cross-feeding mutualism. PLoS Biol. 2016, 14, e1002540. [Google Scholar] [CrossRef] [PubMed]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Dillon, J. Microbial biofilms in the human gastrointestinal tract. J. Appl. Microbiol. 2007, 102, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Donelli, G.; Vuotto, C.; Cardines, R.; Mastrantonio, P. Biofilm-growing intestinal anaerobic bacteria. FEMS Immunol. Med. Microbiol. 2012, 65, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Sahoo, S.; Fleming, R.M.; Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Khan, M.T.; Paglia, G.; Rodionov, D.A.; Harmsen, H.J.; Thiele, I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J. Bacteriol. 2014, 196, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Baumler, D.J.; Peplinski, R.G.; Reed, J.L.; Glasner, J.D.; Perna, N.T. The evolution of metabolic networks of E. coli. BMC Syst. Biol. 2011, 5, 182. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.R.; Shin, J.H.; Cho, J.S.; Yang, D.; Lee, S.Y. Systems Metabolic Engineering of Escherichia coli. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mimee, M.; Tucker, A.C.; Voigt, C.A.; Lu, T.K. Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 2015, 1, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gomez, J.A.; Höffner, K.; Phalak, P.; Barton, P.I.; Henson, M.A. Spatiotemporal modeling of microbial metabolism. BMC Syst. Biol. 2016, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Phalak, P.; Chen, J.; Carlson, R.P.; Henson, M.A. Metabolic modeling of a chronic wound biofilm consortium predicts spatial partitioning of bacterial species. BMC Syst. Biol. 2016, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Lackner, S. Modeling of biofilm systems: A review. In Productive Biofilms; Springer: New York, NY, USA, 2014; pp. 53–76. [Google Scholar]
- Stewart, P.S. Diffusion in biofilms. J. Bacteriol. 2003, 185, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Meadows, A.L.; Karnik, R.; Lam, H.; Forestell, S.; Snedecor, B. Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab. Eng. 2010, 12, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.; Hoffner, K.; Barton, P.I. DFBAlab: A fast and reliable MATLAB code for dynamic flux balance analysis. BMC Bioinform. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Parameter | Value | Units | Source |
|---|---|---|---|---|
| L | Biofilm thickness | 40 | μm | [13] |
| Biomass diffusion coefficients | 1 × 10−10 | cm2/s | Specified | |
| Biomass mass transfer coefficients | 1 × 10−7 | cm/s | Specified | |
| Biomass bulk concentrations | 0 | g/L | Specified | |
| Nutrient diffusion coefficients | 2 × 10−6 | cm2/s | [69] | |
| Nutrient mass transfer coefficients | 2 × 10−4 | cm/s | Specified | |
| Glucose bulk concentration | 5 | mmol/L | Specified | |
| Amino acid bulk concentrations | 0.5 | mmol/L | Specified | |
| Glucose bulk concentration | 0 | mmol/L | Specified | |
| Amino acid bulk concentrations | 0 | mmol/L | Specified | |
| Byproduct diffusion coefficients | 2 × 10−6 | cm2/s | [69] | |
| Byproduct mass transfer coefficients | 5 × 10−6 | cm/s | Specified | |
| Byproduct bulk concentrations | 0 | mmol/L | Specified | |
| Byproduct bulk concentrations | 0 | mmol/L | Specified | |
| Glucose maximum uptake rate | 10 | mmol/gDW/h | [70] | |
| Amino acid maximum uptake rates | 1 | mmol/gDW/h | [70] | |
| Byproduct maximum uptake rates | 10 | mmol/gDW/h | Specified | |
| Michaelis–Menten constants | 0.5 | mmol/L | [70] | |
| Initial biomass concentrations | 10 | g/L | Specified |
| Level | B. thetaiotaomicron | F. prausnitzii | E. coli |
|---|---|---|---|
| 1 | Biomass | Biomass | Biomass |
| 2 | Acetate | Lactate | Acetate |
| 3 | Succinate | Butyrate | Ethanol |
| 4 | Propionate | Formate | Formate |
| 5 | CO2 | CO2 | Lactate |
| 6 | Lactate | Acetate | Succinate |
| 7 | Ethanol | Succinate | CO2 |
| 8 | Formate | Glucose | Butyrate |
| 9 | Glucose | Methionine | Propionate |
| 10 | Methionine | Serine | Glucose |
| 11 | Serine | Tryptophan | Methionine |
| 12 | Tryptophan | Not specified | Serine |
| 13 | Not specified | Not specified | Tryptophan |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henson, M.A.; Phalak, P. Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome. Processes 2017, 5, 13. https://doi.org/10.3390/pr5010013
Henson MA, Phalak P. Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome. Processes. 2017; 5(1):13. https://doi.org/10.3390/pr5010013
Chicago/Turabian StyleHenson, Michael A., and Poonam Phalak. 2017. "Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome" Processes 5, no. 1: 13. https://doi.org/10.3390/pr5010013
APA StyleHenson, M. A., & Phalak, P. (2017). Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome. Processes, 5(1), 13. https://doi.org/10.3390/pr5010013