Organic Polymers as Porogenic Structure Matrices for Mesoporous Alumina and Magnesia




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
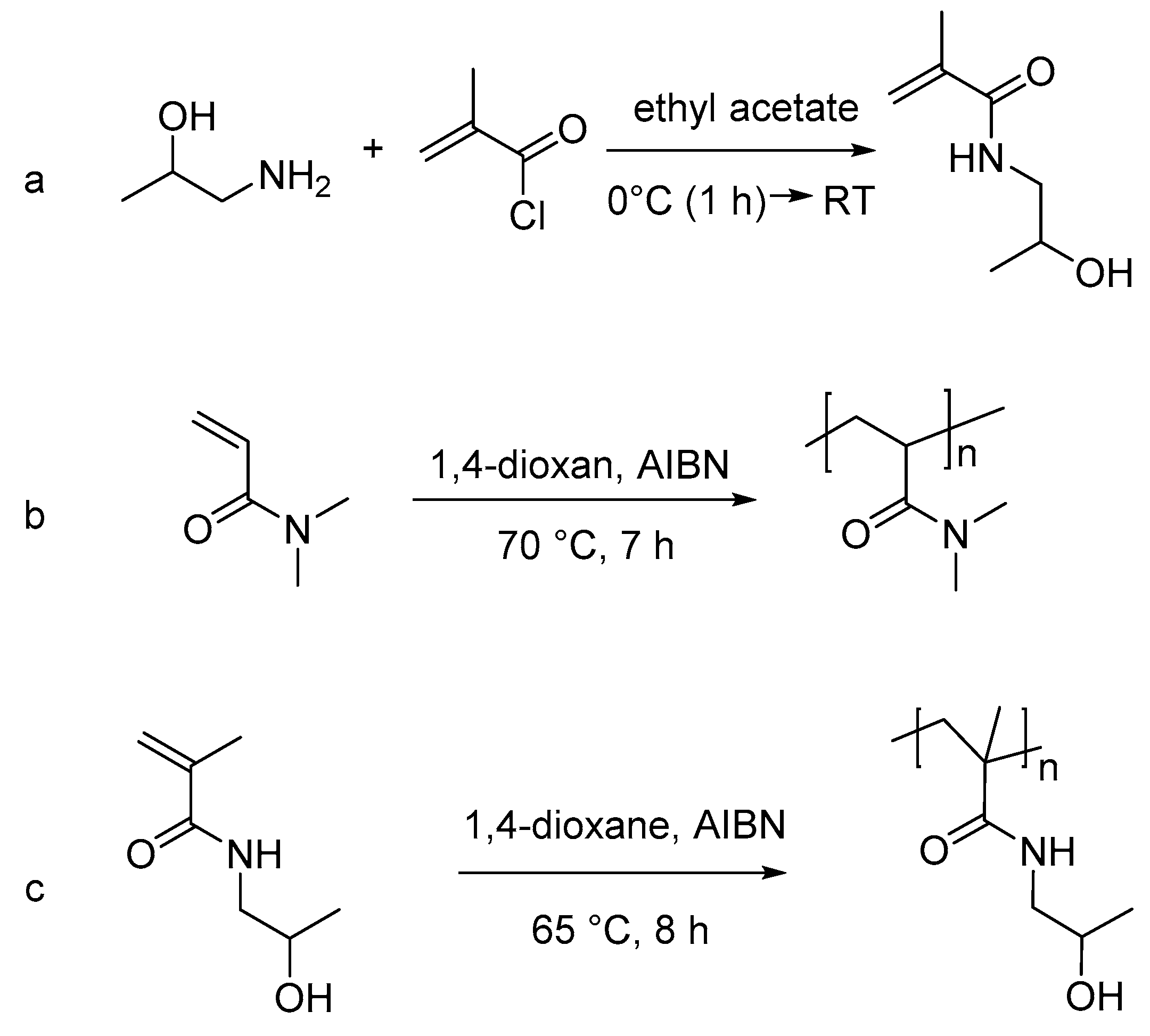
2.3. Monomer Synthesis
2.4. Polymer Synthesis
2.5. Preparation of Mesoporous Metal Oxides
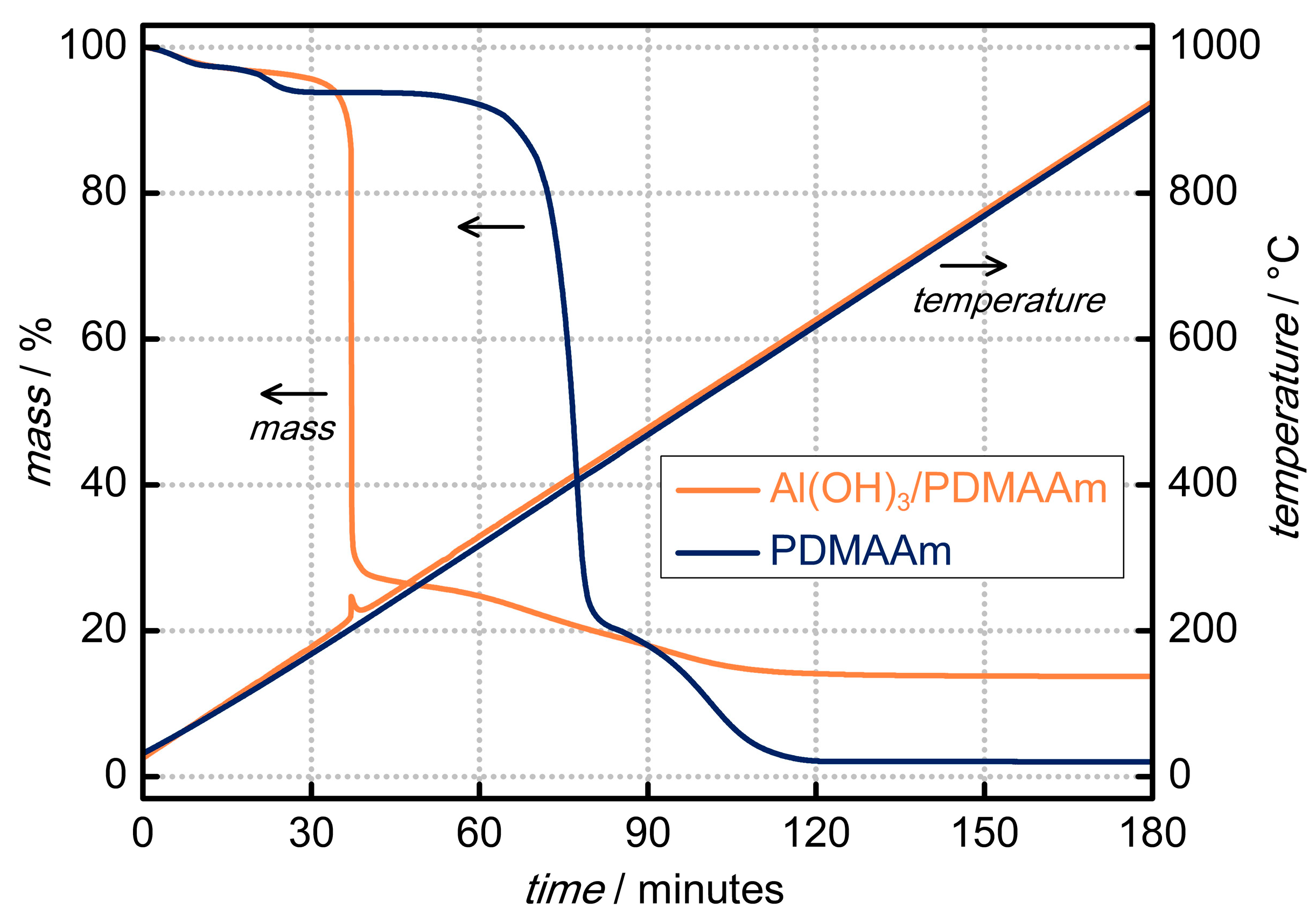
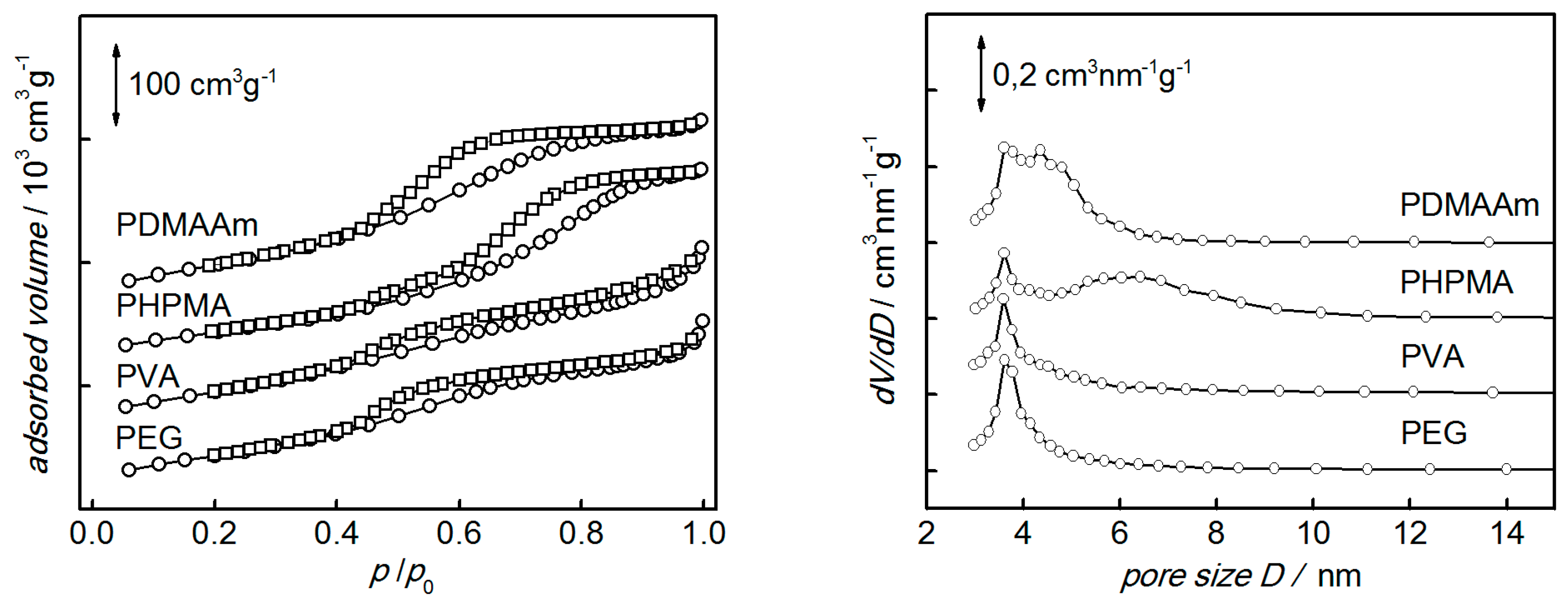
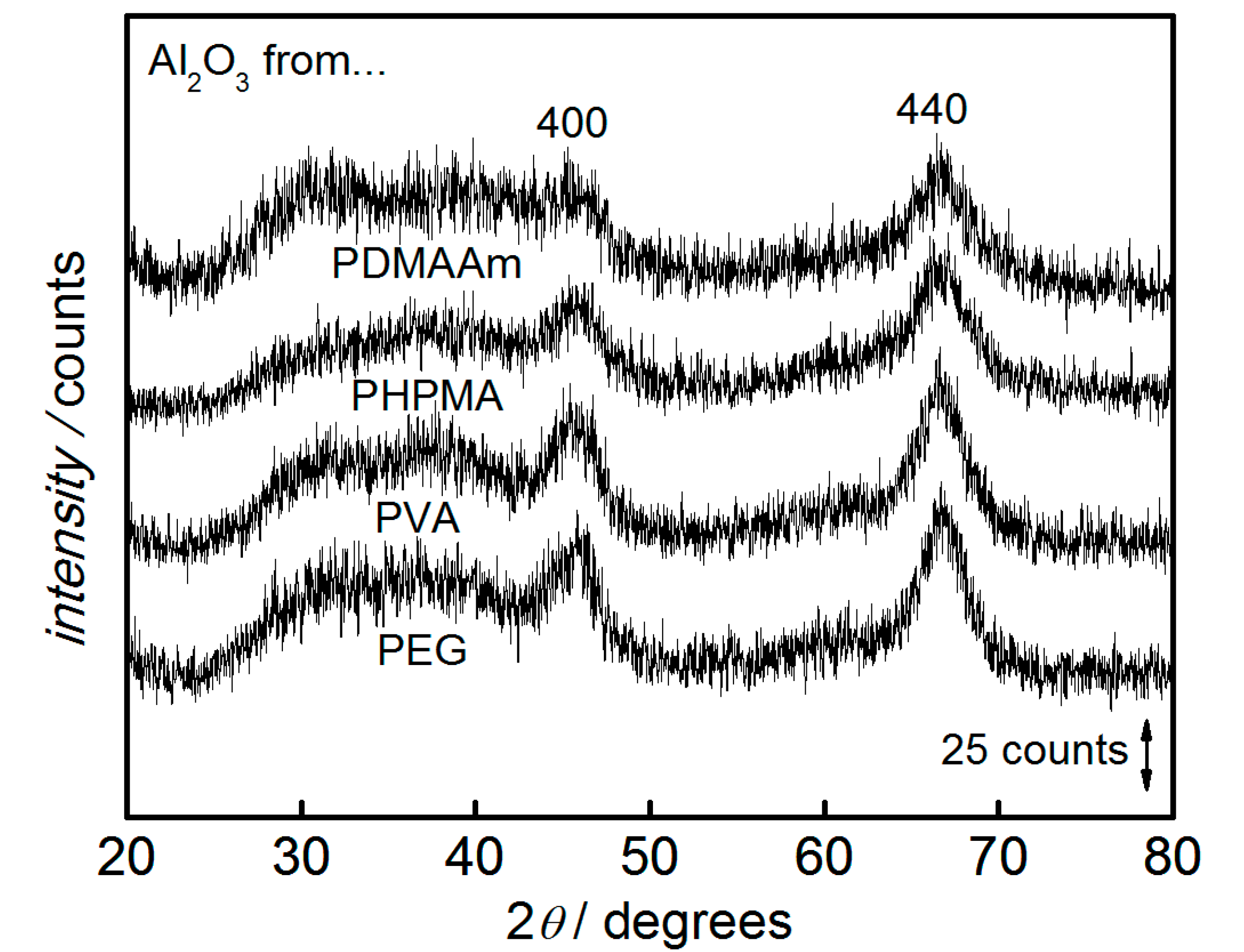
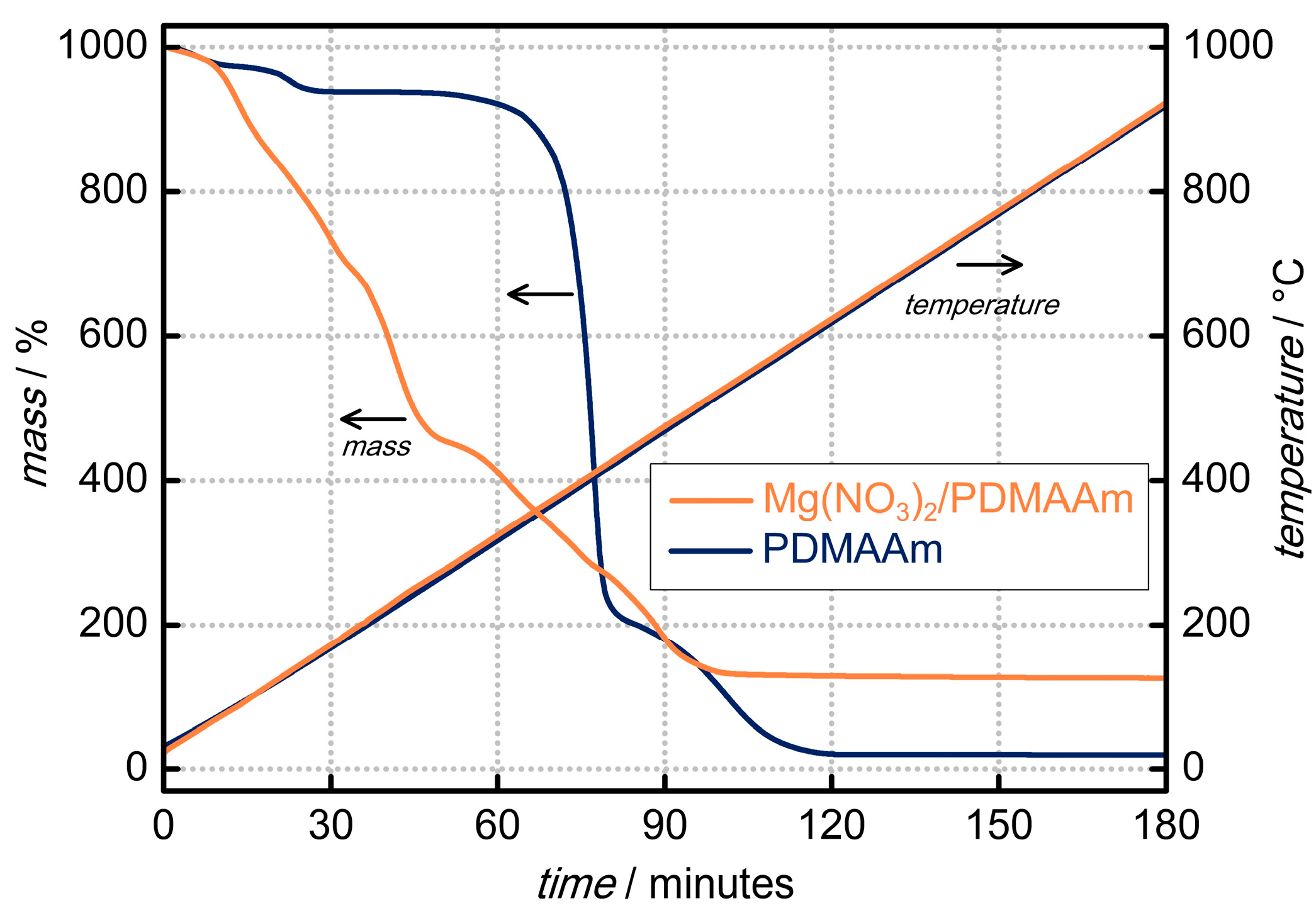
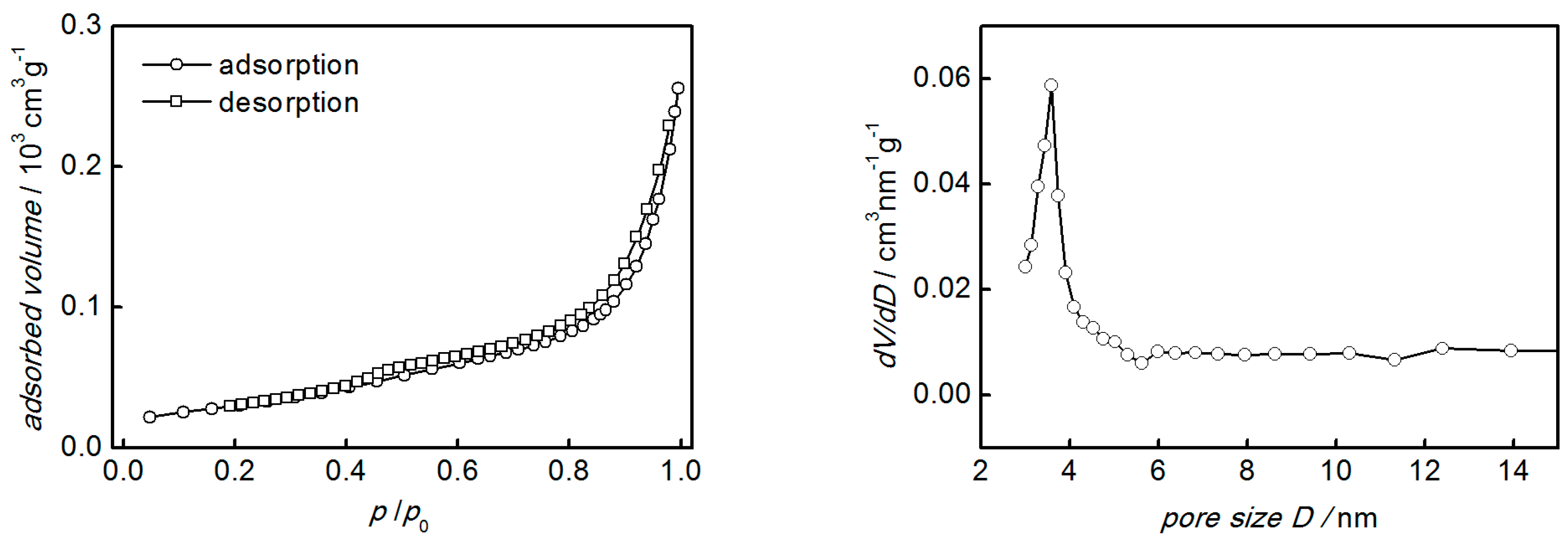
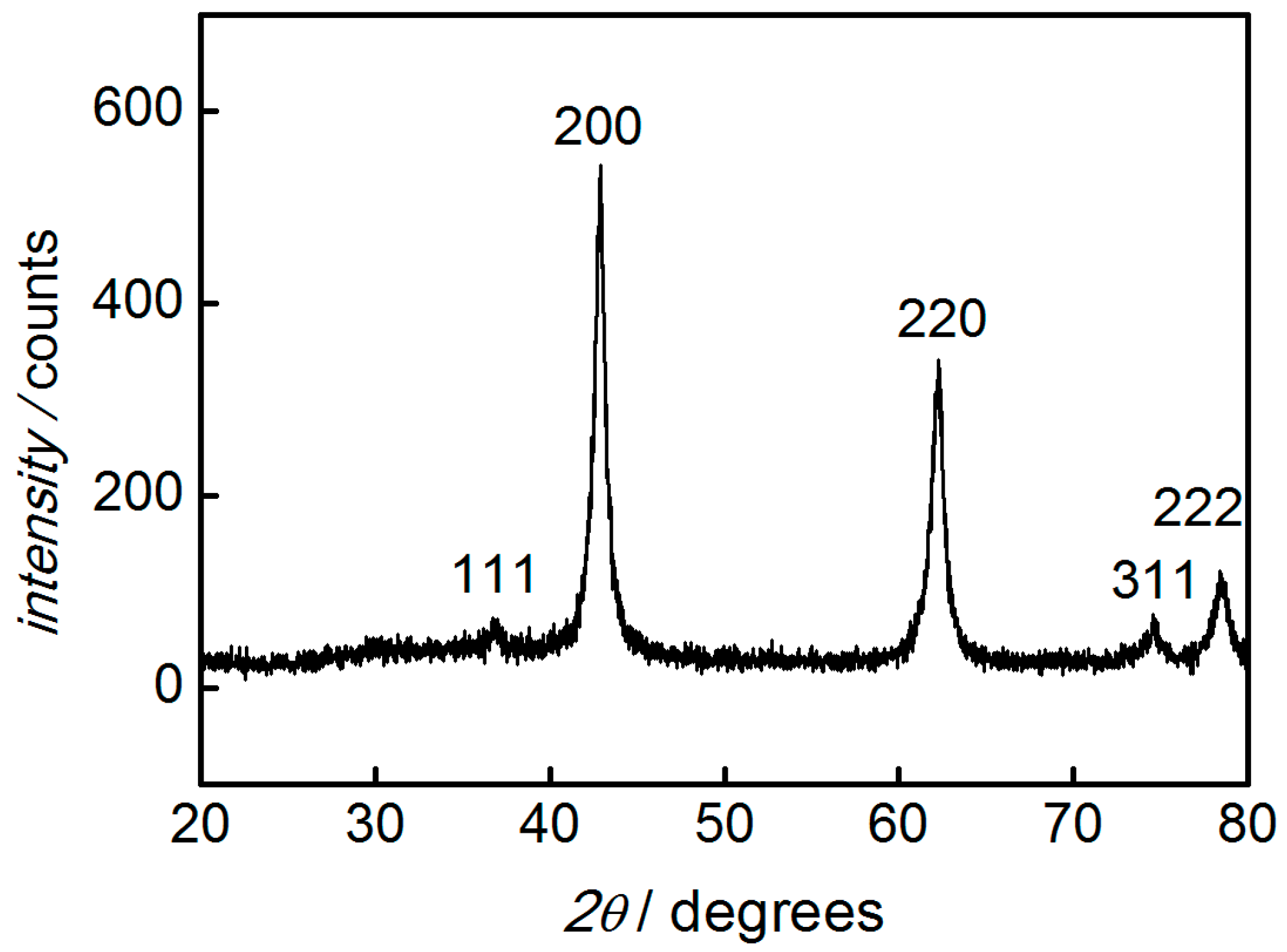
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tüysüz, H.; Schüth, F. Ordered Mesoporous Materials as Catalysts. Adv. Catal. 2012, 55, 127–239. [Google Scholar]
- Li, W.; Liu, J.; Zhao, D. Mesoporous materials for energy conversion and storage devices. Nat. Rev. Mater. 2016, 1, 16023. [Google Scholar] [CrossRef]
- Tiemann, M. Porous metal oxides as gas sensors. Chem. Eur. J. 2007, 13, 8376–8388. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Haffer, S.; Weinberger, C.; Klaus, D.; Tiemann, M. Mesoporous materials as gas sensors. Chem. Soc. Rev. 2013, 42, 4036–4053. [Google Scholar] [CrossRef] [PubMed]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Gu, D.; Schüth, F. Synthesis of non-siliceous mesoporous oxides. Chem. Soc. Rev. 2014, 43, 313–344. [Google Scholar] [PubMed]
- Tiemann, M. Repeated templating. Chem. Mater. 2007, 20, 961–971. [Google Scholar] [CrossRef]
- Ren, Y.; Ma, Z.; Bruce, P.G. Ordered mesoporous metal oxides: Synthesis and applications. Chem. Soc. Rev. 2012, 41, 4909–4927. [Google Scholar] [PubMed]
- Deng, X.; Chen, K.; Tüysüz, H. Protocol for the nanocasting method: Preparation of ordered mesoporous metal oxides. Chem. Mater. 2017, 29, 40–51. [Google Scholar]
- Čejka, J. Organized mesoporous alumina: Synthesis, structure and potential in catalysis. Appl. Catal. A Gen. 2003, 254, 327–338. [Google Scholar]
- Choudary, B.M.; Mulukutla, R.S.; Klabunde, K.J. Benzylation of aromatic compounds with different crystallites of MgO. J. Am. Chem. Soc. 2003, 125, 2020–2021. [Google Scholar] [CrossRef] [PubMed]
- Trueba, M.; Trasatti, S.P. γ-Alumina as a support for catalysts: A review of fundamental aspects. Eur. J. Inorg. Chem. 2005, 17, 3393–3403. [Google Scholar] [CrossRef]
- Morris, S.M.; Fulvio, P.F.; Jaroniec, M. Ordered mesoporous alumina-supported metal oxides. J. Am. Chem. Soc. 2008, 130, 15210–15216. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Koper, O.; Decker, S.; Klabunde, K.J. Nanocrystalline metal oxides as destructive adsorbents for organophosphorus compounds at ambient temperatures. Chem. Eur. J. 2002, 8, 2602–2607. [Google Scholar] [CrossRef]
- Li, L.; Wen, X.; Fu, X.; Wang, F.; Zhao, N.; Xiao, F.; Wie, W.; Sun, Y. MgO/Al2O3 Sorbent for CO2 Capture. Energy Fuels 2010, 24, 5773–5780. [Google Scholar] [CrossRef]
- Wie, J.; Ren, Y.; Luo, W.; Sun, Z.; Cheng, X.; Li, Y.; Deng, Y.; Elzatahry, A.A.; Al-Dahyan, D.; Zhao, D. Ordered mesoporous alumina with ultra-large pores as an efficient absorbent for selective bioenrichment. Chem. Mater. 2017, 29, 2211–2217. [Google Scholar]
- Liu, Q.; Wang, A.; Wang, X.; Zhang, T. Ordered crystalline alumina molecular sieves synthesized via a nanocasting route. Chem. Mater. 2006, 18, 5153–5155. [Google Scholar] [CrossRef]
- Haffer, S.; Weinberger, C.; Tiemann, M. Mesoporous Al2O3 by nanocasting: Relationship between crystallinity and mesoscopic order. Eur. J. Inorg. Chem. 2012, 2012, 3283–3288. [Google Scholar] [CrossRef]
- Roggenbuck, J.; Tiemann, M. Ordered mesoporous magnesium oxide with high thermal stability synthesized by exotemplating using CMK-3 carbon. J. Am. Chem. Soc. 2005, 127, 1096–1097. [Google Scholar] [CrossRef] [PubMed]
- Roggenbuck, J.; Koch, G.; Tiemann, M. Synthesis of mesoporous magnesium oxide by CMK-3 carbon structure replication. Chem. Mater. 2006, 18, 4151–4156. [Google Scholar] [CrossRef]
- Waitz, T.; Tiemann, M.; Klar, P.J.; Sann, J.; Stehr, J.; Meyer, B.K. Crystalline ZnO with an enhanced surface area obtained by nanocasting. Appl. Phys. Lett. 2007, 90, 123108. [Google Scholar] [CrossRef]
- Wagner, T.; Waitz, T.; Roggenbuck, J.; Fröba, M.; Kohl, C.-D.; Tiemann, M. Ordered mesoporous ZnO for gas sensing. Thin Solid Films 2007, 515, 8360–8363. [Google Scholar] [CrossRef]
- Chernikov, A.; Horst, S.; Waitz, T.; Tiemann, M.; Chatterjee, S. Photoluminescence properties of ordered mesoporous ZnO. J. Phys. Chem. C 2011, 115, 1375–1379. [Google Scholar] [CrossRef]
- Polarz, S.; Orlov, A.V.; Schüth, F.; Lu, A.-H. Preparation of High-Surface-Area Zinc Oxide with Ordered Porosity, Different Pore Sizes, and Nanocrystalline Walls. Chem. Eur. J. 2007, 13, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Roggenbuck, J.; Waitz, T.; Tiemann, M. Synthesis of Mesoporous Metal Oxides by Structure Replication: Strategies of Impregnating Porous Matrices with Metal Salts. Microporous Mesoporous Mater. 2008, 113, 575–582. [Google Scholar] [CrossRef]
- Weinberger, C.; Roggenbuck, J.; Hanss, J.; Tiemann, M. Synthesis of Mesoporous Metal Oxides by Structure Replication: Thermal Analysis of Metal Nitrates in Porous Carbon Matrices. Nanomaterials 2015, 5, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Llusar, M.; Pidol, L.; Roux, C.; Pozzo, J.L.; Sanchez, C. Templated Growth of Alumina-Based Fibers through the Use of Anthracenic Organogelators. Chem. Mater. 2002, 14, 5124–5133. [Google Scholar] [CrossRef]
- Jiu, J.; Kurumada, K.; Tanigaki, M. Preparation of oxide with nano-scaled pore diameters using gel template. J. Non-Cryst. Solids 2003, 325, 124–132. [Google Scholar] [CrossRef]
- Kurumada, K.; Suzuki, A.; Baba, S.; Otsuka, E. Relationship between polarity of template hydrogel and nanoporous structure replicated in sol–gel-derived silica matrix. J. Appl. Polym. Sci. 2009, 114, 4085–4090. [Google Scholar] [CrossRef]
- Cui, X.; Tang, S.; Zhou, H. Mesoporous alumina materials synthesized in different gel templates. Mater. Lett. 2013, 98, 116–119. [Google Scholar] [CrossRef]
- Jiang, R.; Zhu, H.-Y.; Chen, H.-H.; Yao, J.; Fu, Y.-Q.; Zhang, Z.-Y.; Xu, Y.-M. Effect of calcination temperature on physical parameters and photocatalytic activity of mesoporous titania spheres using chitosan/poly(vinyl alcohol) hydrogel beads as a template. Appl. Surf. Sci. 2014, 319, 189–196. [Google Scholar] [CrossRef]
- Kuckling, D.; Hoffmann, J.; Plötner, M.; Ferse, D.; Kretschmer, K.; Adler, H.-J.P.; Arndt, K.-F.; Reichelt, R. Photo cross-linkable poly(N-isopropylacrylamide) copolymers III: micro-fabricated temperature responsive hydrogels. Polymer 2003, 44, 4455–4462. [Google Scholar] [CrossRef]
- Döring, A.; Birnbaum, W.; Kuckling, D. Responsive hydrogels—structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 40, 7391–7420. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, W.; Weinberger, C.; Schill, V.; Haffer, S.; Tiemann, M.; Kuckling, D. Synthesis of mesoporous alumina through photo cross-linked poly(dimethylacrylamide) hydrogels. Colloid Polym. Sci. 2014, 292, 3055–3060. [Google Scholar] [CrossRef]
- Weinberger, C.; Chen, Z.; Birnbaum, W.; Kuckling, D.; Tiemann, M. Photo-cross-linked polydimethylacrylamide hydrogels as porogens for mesoporous alumina. Eur. J. Inorg. Chem. 2017, 2017, 1026–1031. [Google Scholar] [CrossRef]
- Cottet, H.; Gareil, P. Electrophoretic behavior of fully sulfonated polystyrenes in capillaries filled with entangled polymer solutions. J. Chromatogr. Coruña 1997, 772, 369–384. [Google Scholar] [CrossRef]
- Daoud, M.; Stanley, H.E.; Stauffer, D. Scaling, Exponents, and Fractal Dimensions. In Physical Properties of Polymers Handbook, 2nd ed.; Mark, J.E., Ed.; Springer: New York, NY, USA, 2007; pp. 83–92. [Google Scholar]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Rowe, M.D.; Chang, C.C.; Thamm, D.H.; Kraft, S.L.; Harmon, J.F.; Vogt, A.P.; Sumerlin, B.S.; Boyes, S.G. Tuning the magnetic resonance imaging properties of positive contrast agent nanoparticles by surface modification with RAFT polymers. Langmuir 2009, 25, 9487–9499. [Google Scholar] [CrossRef] [PubMed]
- Kuckling, D.; Harmon, M.E.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 1. Synthesis and characterization of constrained temperature-responsive hydrogel layers. Macromolecules 2002, 35, 6377–6383. [Google Scholar] [CrossRef]
- Javadi, A. Synthesis of Thin Hydrogel Layers Based on Photo-Cross-Linkable Polymers. Ph.D. Thesis, Tarbiat Moallem University, Tehran, Iran, May 2012. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn/(g mol−1) | D | Yield/% |
|---|---|---|---|
| PEG 1 | 12,000 | 1.1 | - |
| PVA 1 | 23,000 | 2.4 | - |
| PDMAAm 1 | 26,000 | 2.8 | 68 |
| PHPMA 1 | 43,000 | 6.3 | 77 |
| Polymer Used | ABET/m2 g−1 | V/cm3 g−1 | r/nm |
|---|---|---|---|
| PDMAAm | 365 | 0.51 | 3.6 |
| PHPMA | 312 | 0.54 | 3.6 |
| PEG | 325 | 0.44 | 3.6 |
| PVA | 343 | 0.48 | 3.6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Weinberger, C.; Tiemann, M.; Kuckling, D. Organic Polymers as Porogenic Structure Matrices for Mesoporous Alumina and Magnesia. Processes 2017, 5, 70. https://doi.org/10.3390/pr5040070
Chen Z, Weinberger C, Tiemann M, Kuckling D. Organic Polymers as Porogenic Structure Matrices for Mesoporous Alumina and Magnesia. Processes. 2017; 5(4):70. https://doi.org/10.3390/pr5040070
Chicago/Turabian StyleChen, Zimei, Christian Weinberger, Michael Tiemann, and Dirk Kuckling. 2017. "Organic Polymers as Porogenic Structure Matrices for Mesoporous Alumina and Magnesia" Processes 5, no. 4: 70. https://doi.org/10.3390/pr5040070
APA StyleChen, Z., Weinberger, C., Tiemann, M., & Kuckling, D. (2017). Organic Polymers as Porogenic Structure Matrices for Mesoporous Alumina and Magnesia. Processes, 5(4), 70. https://doi.org/10.3390/pr5040070