1. Introduction
The in silico study of metabolism has largely transitioned from a specialty discipline to a mainstream biological approach due to improvements in software usability, increases in computational power, and the accumulation of omics databases. Cellular growth is an essential component of many of these computational biology studies [
1,
2,
3]. Understanding the foundation of growth from the level of mass and energy fluxes remains critical for interpretation and integration of in silico metabolic models and omics datasets. The macromolecular composition of a cell is one such area of basic knowledge. Macromolecular composition of both prokaryotic and eukaryotic cells is governed by allocation of resources and can shift depending on cell cycle, specific growth rate, and diel cycle (e.g., cyanobacteria and green algae) [
4,
5,
6].
Stoichiometric modeling approaches analyze steady state fluxes based on metabolic reactions identified from an organism’s genomic potential, enzyme-coding genes identified in the genome sequence [
7]. These methods can be applied to microbial communities as well as individual species [
8,
9]. Optimal metabolic pathways are often assessed in terms of growth: constraint-based approaches, such as flux balance analysis [
10], typically use production of biomass as an objective function, and macromolecular composition dictates the metabolic precursors necessary for growth. Different weightings of macromolecular components in the biomass synthesis reaction can influence results by shifting requirements for precursors [
11]. However, the proportions of biomass components are not specified by the genome sequence [
12]. While technologies for automatic model construction are rapidly increasing, stoichiometric coefficients for the biomass reaction are still necessary [
13]. Often, coefficients for this essential reaction are borrowed from literature reported for
Escherichia coli or an organism similar in physiology or phylogeny to the organism being modeled (e.g., [
14,
15]). However, these values may not be representative of the organism under study. In biotechnology applications, a specific macromolecular component may be targeted, such as lipids extracted for biofuels [
16] or starch compounds for biochemical production. Accurate quantification of these components is important for comparison of production potential under different conditions. Additionally, ratios of macromolecule pools, such as protein, DNA, or RNA, from a microbial population can be correlated to important culture properties, including specific growth rate [
17].
A variety of methods for quantification of any given macromolecule can be found in the literature (e.g., [
18]). Many of these methods date back several decades, and numerous adaptations have arisen over the years. Selecting and implementing a method with an assurance of valid and accurate results relevant to computational biology applications can present a significant challenge, particularly when testing new organisms. Additionally, not all reported methods have been developed for or tested on prokaryotes, and different organisms may respond differently to treatment conditions. For example, cell wall type may influence the efficacy of reagents or procedures, resulting in a method with varying degrees of efficiency for different types of microorganisms. External factors, such as materials used, can also affect the outcome of an analysis, and specific procedural details not included in publications can hinder reproducibility. Recently, methods for determining multiple biomass components with a single technique, e.g., gas chromatography-mass spectrometry, have been developed [
19] but still rely on adequate cell lysis techniques and standard compounds for quantification. A concise collection of information about the variety of existing methods for each macromolecule, including advantages and disadvantages of methods, specific procedural details, and points for potential pitfalls, is a useful resource that is lacking from the published literature.
The current work fills this gap with objectives: (1) to review and compare existing literature regarding methods to measure five major macromolecules (carbohydrate, DNA, lipid, protein, and RNA); (2) to develop a select step-by-step protocol for each macromolecule and test the efficacy on different types of bacterial samples; and (3) to demonstrate the application to computational biology by generating biomass synthesis reactions. Three bacterial species were used as test cases in the current work: E. coli (Gram-negative, mesophilic model laboratory organism), Synechococcus sp. PCC 7002 (Gram-negative, mesophilic cyanobacterium; Synechococcus 7002 hereafter), and Alicyclobacillus acidocaldarius (Gram-positive, thermophilic acidophile). These microorganisms encompass a range of physiological capabilities and characteristics, including photosynthesis and alicyclic fatty acids. The impact of biomass composition on model predictions was demonstrated using essential parameters, including biomass yield on electron donor, biomass yield on electron acceptor, biomass yield on nitrogen, biomass degree of reduction, and growth associated maintenance energy. The results highlight the importance of appropriate methods for the accurate determination of macromolecule composition. Compiling a literature review in conjunction with laboratory-tested protocols with demonstrated application to metabolic models, all within a single source, serves as a useful resource for the computational biology community that should facilitate model building transparency and reproducibility.
3. Modeling Methods
A metabolic network model for
A. acidocaldarius was constructed in CellNetAnalyzer [
26,
27] from the annotated genome [
28] with the aid of MetaCyc, KEGG, BRENDA, and NCBI [
29,
30,
31] databases. Reversible exchange reactions were defined for protons and water. Irreversible exchange reactions were defined to permit ammonium, sulfate, oxygen, and glucose or xylose uptake and carbon dioxide evolution, as well as secretion of possible byproducts, including acetate, lactate, ethanol, and formate. Macromolecular synthesis reactions were defined for nucleic acids, glycogen, lipid, and protein. Synthesis reactions utilized two phosphate bonds per nucleic acid monomer, one phosphate bond per glycogen monomer, and four phosphate bonds per protein monomer [
32]. Nucleotide distributions were set based on percent GC content of the genome for DNA and nucleotide sequence of the rRNA genes for RNA. Fatty acid distribution was assigned based on literature values [
33,
34]. The amino acid distribution was set using the experimentally measured values in the current study. All reactions were balanced for elements, charge, and electrons. Thermodynamic considerations were built into the model via reaction reversibilities based on data from BRENDA [
31]. Model simulations were performed with elementary flux mode analysis. Flux vectors v satisfying the stoichiometric matrix S at steady state (Sv = 0) subject to conservation of mass, specified irreversibilities, and indecomposability constraints were computed, resulting in the collection of minimal pathways through the network, called elementary flux modes (EFMs) [
35]. EFMs were enumerated using EFMtool [
36]. Analysis of resulting EFMs (e.g., biomass yield) was performed with MATLAB. Maintenance energy was fit to experimental glucose and oxygen yield data for
A. acidocaldarius obtained from [
37]. Both growth associated (dominant in fast-growing environmental conditions) and non-growth associated (dominant in slow-growing environmental conditions) maintenance terms were determined. The metabolic model with supporting details, CellNetAnalyzer metabolite and reaction input, an SBML file, and maintenance calculations can be found in the
Supplementary Materials (Files S1, S2, and S3).
9. Model Biomass Reaction
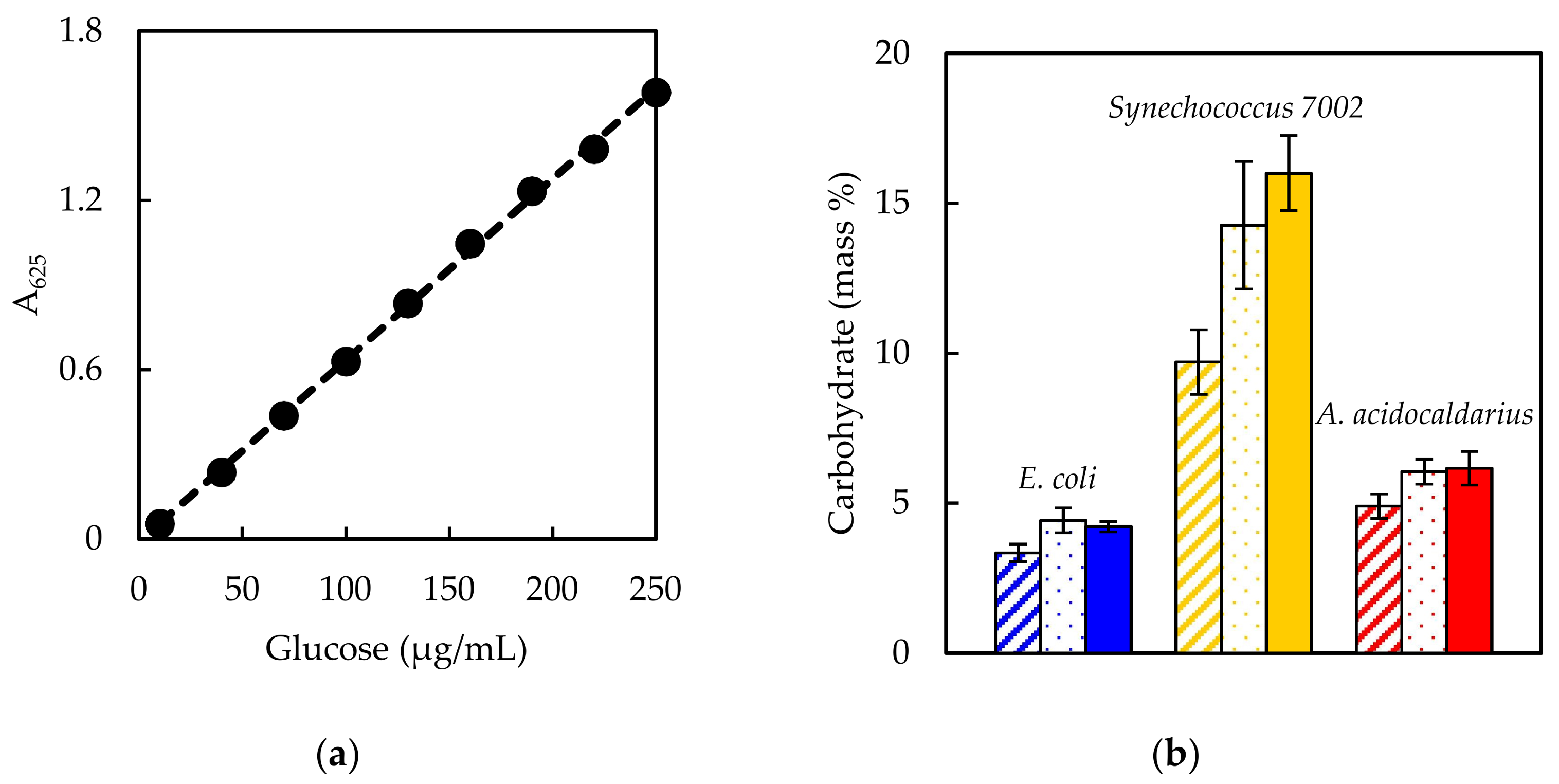
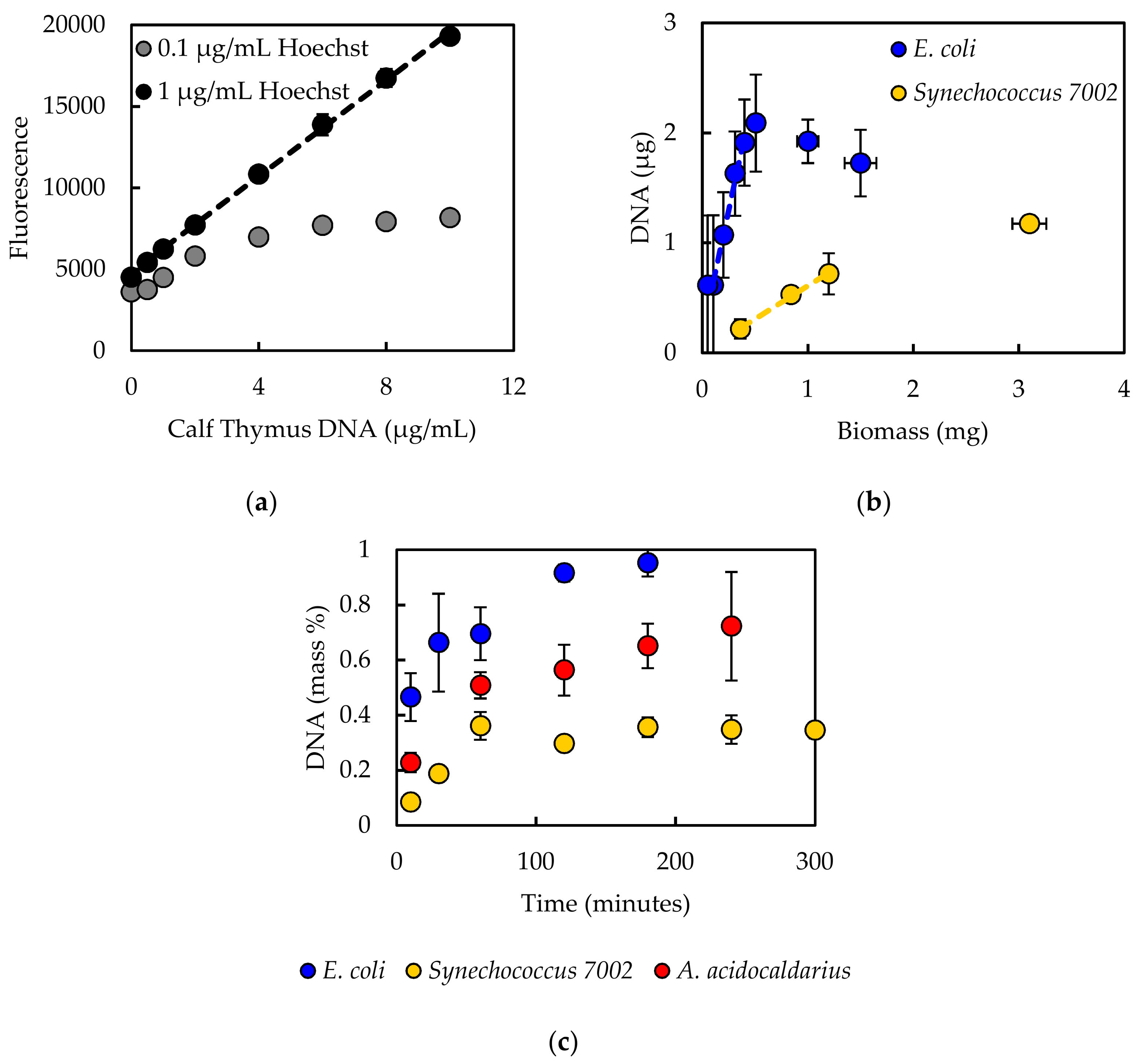
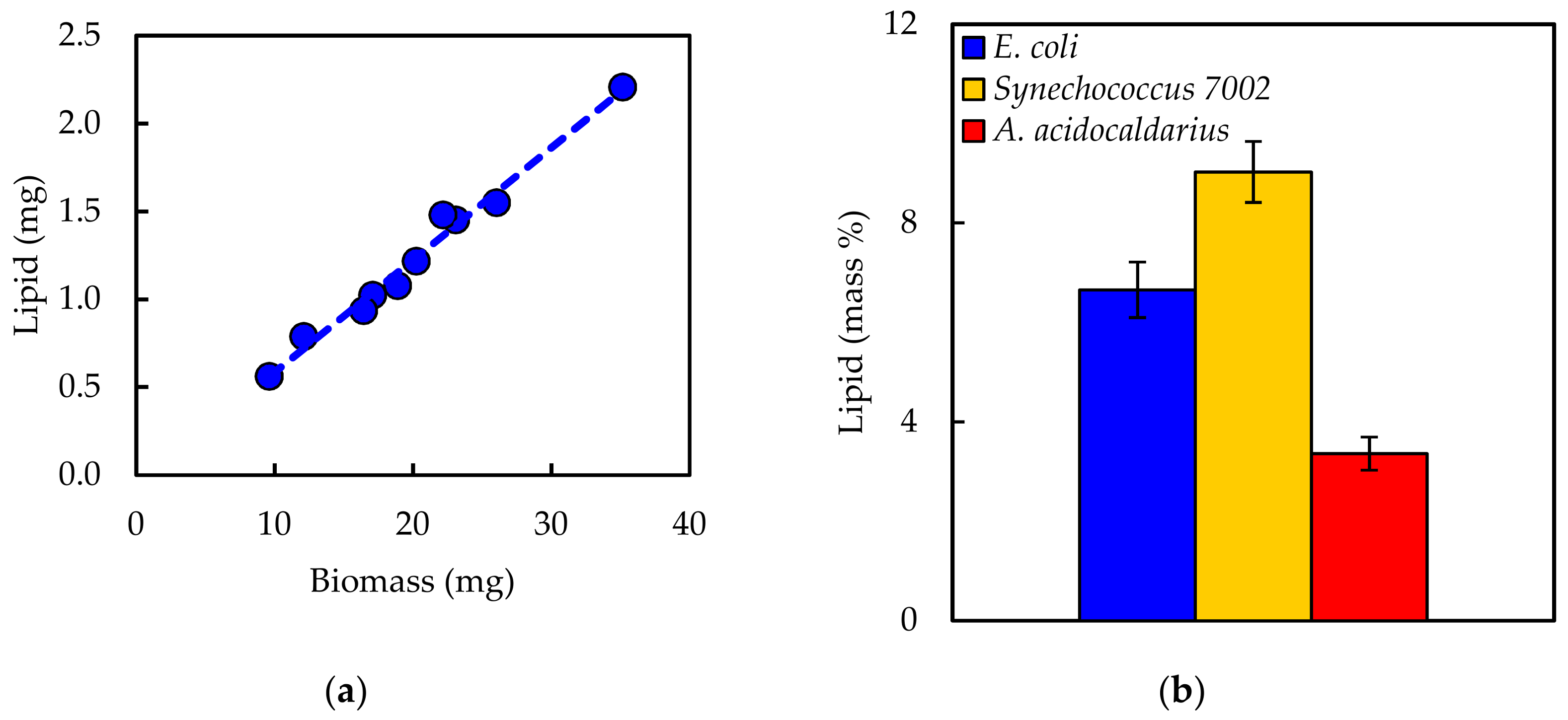
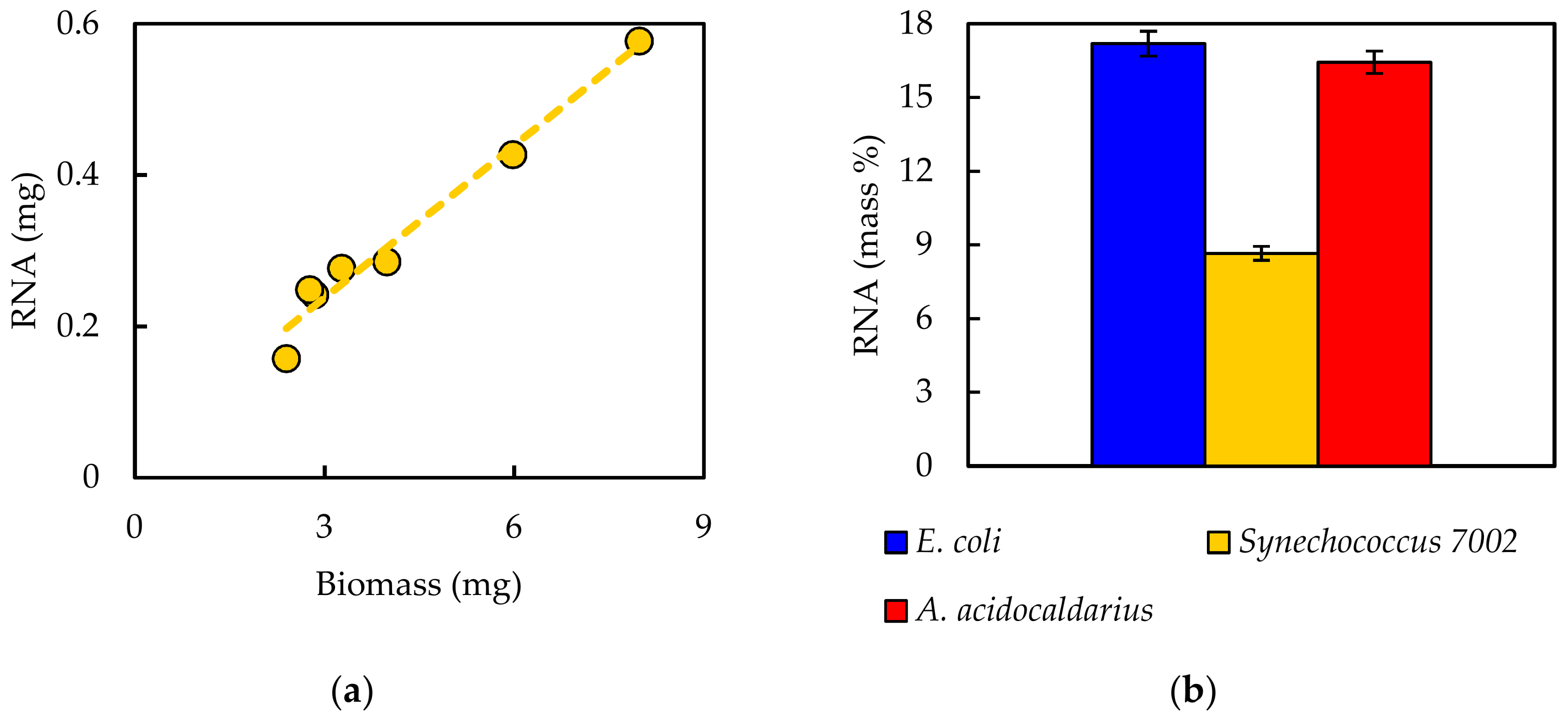
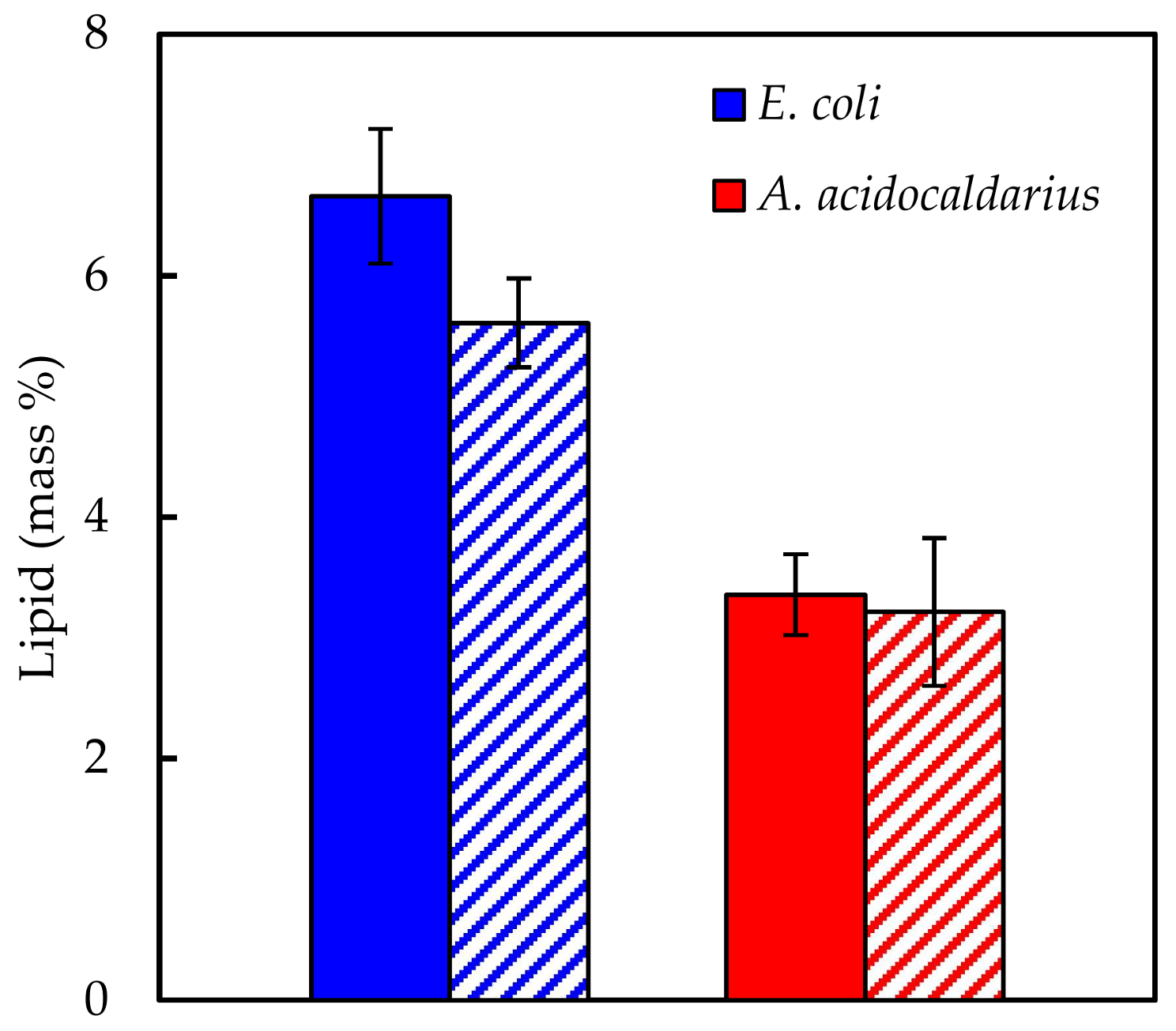
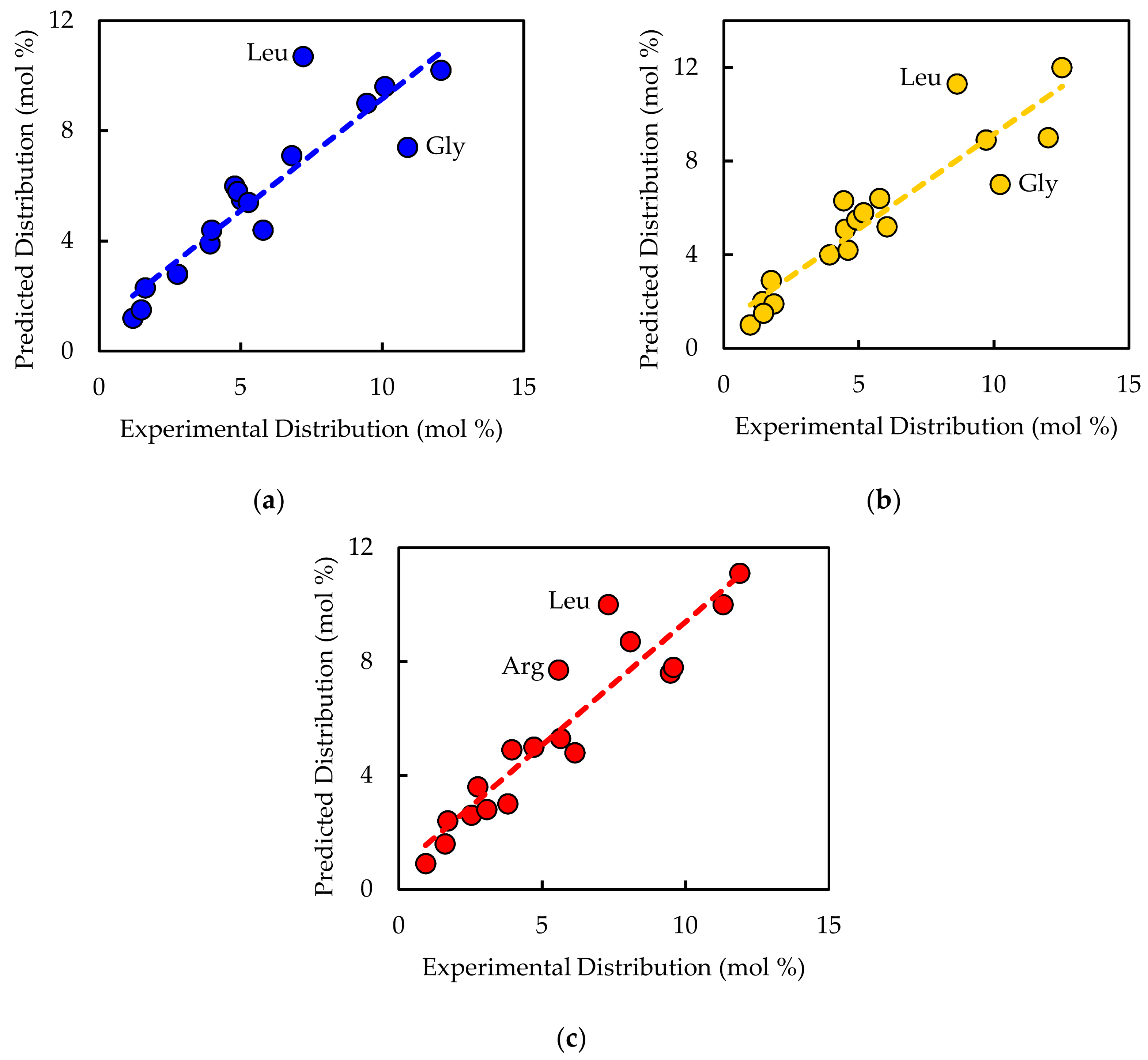
Experimentally measured biomass composition provides a species-relevant basis for representing cellular growth in computational models. The results of the macromolecular assays for
E. coli,
Synechococcus 7002, and
A. acidocaldarius are summarized in
Table 4. The mass percentages for the five assays do not necessarily sum to 100% of cell dry weight. The reduced mass recovery may be due to loss of biomass during centrifugation and transfer of material while performing the assays. Some bacteria may also possess other storage compounds that are not accounted for in these analyses, such as polyhydroxyalkanoates or polyphosphates. Ash weight typically accounts for 5–10% of cell dry weight [
72], or perhaps even more for some organisms (e.g., 20–30% ash content has been measured in phytoplankton [
73]). To adjust for losses during sample processing, measurements can be normalized to the total mass recovered such that the sum of biomass recovered from all measurements is 100% (
Table 4).
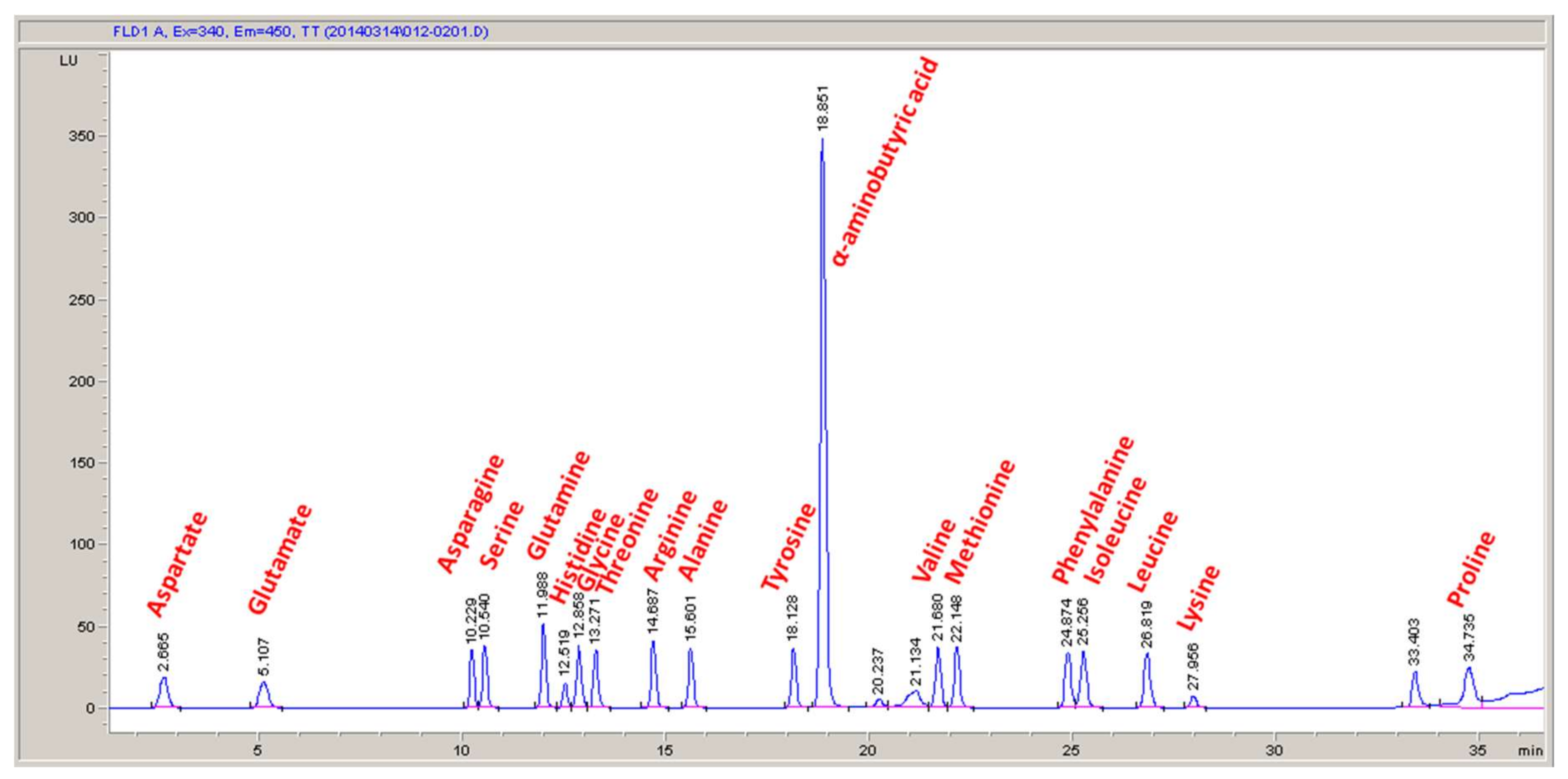
An in silico cellular growth reaction is a collection of macromolecular synthesis reactions scaled to account for biomass composition. The macromolecular synthesis reactions are constructed by accounting for the appropriate ratios of the monomers, polymerization energy requirements, and reaction byproducts. Macromolecular monomer distributions are either measured directly, such as the amino acid composition measured here, or can be estimated from appropriate omics datasets or the literature. DNA composition is typically estimated from GC content, and RNA composition may be estimated from rRNA-encoding genes; rRNA accounts for approximately 81% of cellular RNA [
32]. Polymer lengths for the macromolecular synthesis reactions can be scaled to a convenient number of monomers, such as 10 or 100, with the appropriate polymerization energy requirements and byproducts. The polymerization energy error introduced with these scaled molecules is assumed minor.
Once formulas for individual macromolecules are calculated, model reactions can be quality control checked for balance of elemental formulas and degree of reduction to ensure adherence to the mass balance constraint required for stoichiometric modeling. Identification of imbalanced reactions can then be further investigated; often the issue can be traced to balancing of redox pairs or hydrolysis products, free protons, and water.
Table 5 demonstrates the construction of a DNA macromolecule synthesis reaction for
A. acidocaldarius, including the definition of monomer composition, polymerization energy requirements, and byproducts. The elemental and electron balances are included and validate conservation relationships [
32]. The
Supplementary Materials contain a workbook for the major biomass macromolecules that can be modified for different biomass measurements (
File S4).
The overall cell growth reaction has a form analogous to A carbohydrate + B DNA + C lipid + D protein + E RNA = 1 biomass, where A, B, C, D, and E are stoichiometric coefficients corresponding to the measured mass fraction. Some biomass reactions may also include additional constituents, such as chlorophyll, salts, and metabolite pools, including vitamins. The coefficients for the macromolecular constituents A–E are obtained by converting the experimental mass fraction measurements to molar coefficients, thereby yielding the appropriate stoichiometries. The following steps convert experimentally measured mass fractions of macromolecules to molar coefficients for use in the biomass reaction:
- (1)
Record mass fractions as g macromolecule per g cell dry weight (see
Table 4).
- (2)
Tabulate the molar mass of each macromolecule representation. Multiply the macromolecular formula by the atomic mass of the respective elements, and sum over all elements to obtain g/mol macromolecule.
- (3)
Divide the mass fraction of the macromolecule by its molar mass to obtain mol macromolecule/g cell dry weight. The basis for cell dry weight normalization can be selected as desired; 1, 10, or 100 kg cell dry weight typically results in reasonably scaled coefficients for elementary flux mode and flux balance analyses. One kilogram cell dry weight often provides a convenient basis, as when inputs are scaled to a mM basis in FBA, the resulting output biomass scales to grams.
- (4)
Incorporate the molar coefficients into the biomass reaction. The stoichiometries can be multiplied by the macromolecular formulas and summed over all the macromolecules to obtain an overall formula for biomass, which allows model output to be analyzed in terms of carbon moles of biomass (
Table 6).
The
Supplementary Materials detail the macromolecule and biomass calculations for each species, as well as demonstrate a quality control check for balancing mass, charge, electrons, and elemental composition (
File S4).
In addition to the macromolecular constituents that comprise a cell, metabolic models often account for maintenance energy requirements. Maintenance energy is an implicit energy consumption term accounting for a myriad of cellular processes, such as protein turnover and osmotic pressure maintenance. Maintenance energy is typically estimated by fitting the in silico model to experimental biomass-on-substrate yield data. For example, experiments correlating substrate consumption rate (for heterotrophs) or photon absorption rate (for photoautotrophs) with growth rate can be used to determine the yield [
74,
75]. For elementary flux mode analysis applications, a single maintenance energy term, set for a defined growth rate, can be added to the biomass reaction. For flux balance analysis applications, maintenance energy requirements can be broken down into growth and non-growth associated maintenance (GAM and NGAM) terms. The
Supplementary Materials contain a genome-enabled model constructed for
A. acidocaldarius (
File S1). Calculations fitting maintenance energy to observed yield data for both glucose and oxygen consumption from Farrand et al. [
37] for both EFMA and FBA application are provided in MATLAB and Excel formats (
Files S1, S2, and S3). The specific growth rate-dependent (µ, h
−1) maintenance energy requirement (q
ATP) for
A. acidocaldarius was calculated to be q
ATP = 13.4µ + 4.2 mmol cellular energy per g biomass per hour, where GAM was 13.4 mmol cellular energy (phosphodiester bonds) per g biomass and NGAM was 4.2 mmol cellular energy per g biomass per hour. Using multiple datasets to fit the maintenance energy provides a metric of accuracy for the model, as they should provide similar results. The calculated maintenance terms for
A. acidocaldarius were similar regardless of fitting with glucose or oxygen consumption data (
Files S1, S2, and S3).
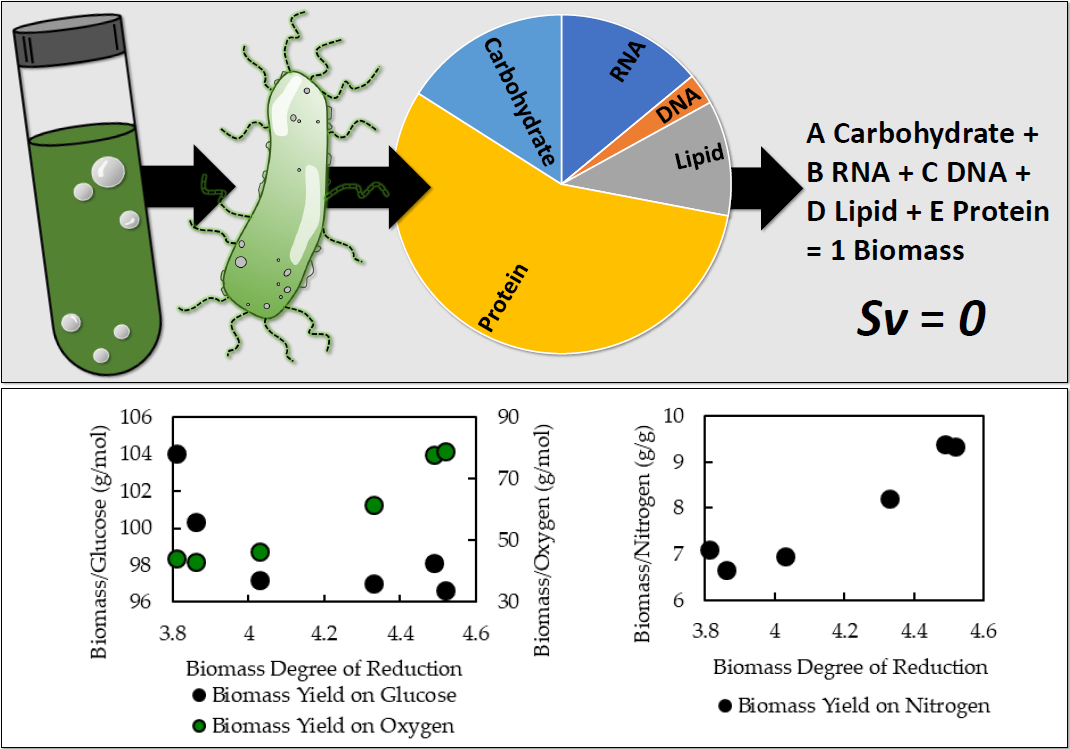
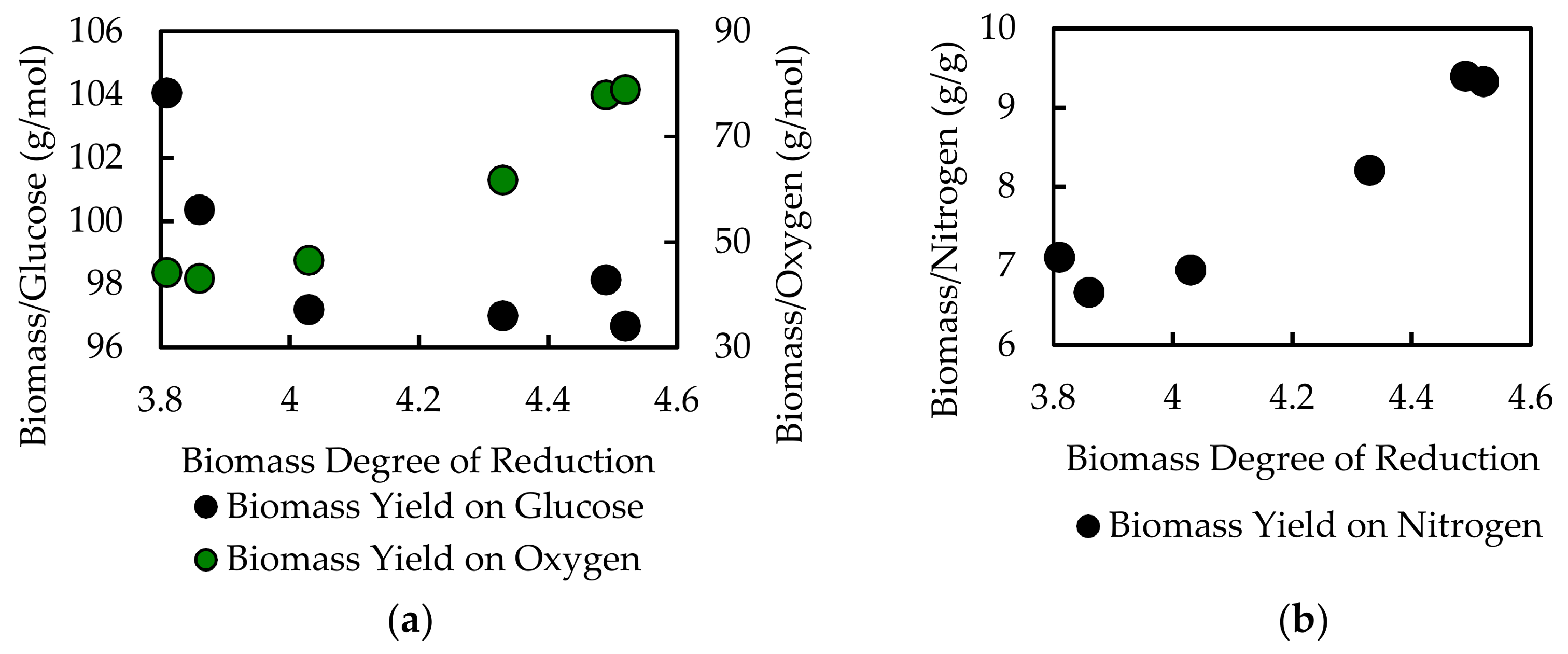
Finally, the
A. acidocaldarius model was used to quantify potential pitfalls associated with inaccurate biomass compositions. Ten different biologically relevant variations of biomass composition were generated and tested in addition to the experimentally measured composition (see
File S5). The optimal in silico biomass yield on electron donor (glucose) and associated biomass yield on electron acceptor (oxygen) was determined for each biomass composition. A sampling of the data is presented as a function of the biomass degree of reduction in
Figure 6 and
Table 7. The data point at degree of reduction 4.03 represents the experimentally measured composition for
A. acidocaldarius; this point is used as a reference. The in silico biomass per glucose and biomass per oxygen yields change nonlinearly relative to degree of reduction. The biomass per oxygen yields change up to 70% from the reference composition, demonstrating the strong influence biomass composition can have on simulation results (
Figure 6,
Table 7). Common modeling practices for determining maintenance energy parameters fit model output to experimental yield data, which can mask the effects of inaccurate biomass composition. GAM values for each biomass composition were also calculated (
Figure 6,
Table 7). The GAM values changed up to 40% over the reference case. This represents a substantial 40% change in specific energy generation-associated fluxes, such as ATPase. Furthermore, the biomass yield on nitrogen was calculated for each biomass composition. The biomass per nitrogen yields varied up to 35% for the considered biomass compositions (see
File S5). This variation in nitrogen content would have substantial impact on predictions for nitrogen-limited culturing conditions, such as those commonly used in bioprocesses to induce accumulation of bioplastics or lipids (e.g., [
76,
77]). This analysis highlights the importance of accurate species- and condition-specific measurements for biomass composition.
10. Conclusions
Computational biology representations of metabolism often include cellular growth reactions necessitating knowledge of biomass composition for accurate predictions. The current work surveyed analytical methods for the five major macromolecules (carbohydrate, DNA, lipid, protein, and RNA), provided step-by-step procedures for a select method for each macromolecule, tested the methods on three different bacterial species, and demonstrated application of analytical measurements to a computational representation of cellular growth. The data include a quantitative analysis of potential pitfalls associated with inaccurate biomass representations. The literature survey included references to more in-depth reviews for each macromolecule for further exploration and also provided a rationale for the selected method.
Table 8 provides a summary of the selected methods and their advantages and disadvantages. The three bacterial species used for testing (
E. coli,
Synechococcus 7002, and
A. acidocaldarius) represent a range of physiological characteristics, including Gram-negative and Gram-positive, mesophilic and thermophilic, and neutrophilic and acidophilic, as well as chemoheterotrophic and photoautotrophic, which assessed the robustness of the methods. Testing of methods highlighted potential pitfalls and provided guidelines for troubleshooting when testing a new method or when applying a method to new organisms. Based on the current study, recommendations for verifying a new protocol or testing a new organism include ensuring that the test response is linear for both the amount of biomass used and the amount of reagent, testing the standard range, and confirming the effect of any sample pre-treatment steps on standards. It is also important to consider the organism being studied and the downstream application of the measurement (e.g., glycogen vs. total carbohydrate).
The presented methods of experimental measurement and conversion to computational biology reactions need to be integrated with the maturing quality standards for model construction [
78,
79]. The predicted elemental composition of the synthesized biomass is a relevant metric for the quality of the overall reaction. Average elemental compositions have been measured for several common microorganisms, providing a convenient check [
80]. The elemental composition is linked to the biomass degree of reduction, which is an energetic measure of biomass and a critical parameter for computational biology analysis of consortia simulations. The degree of reduction of biomass for an average cell is approximately 4.2 or 4.8 on an NH
4+ or N
2 basis, respectively [
80]. These values can shift due to large quantities of cellular storage polymers, such as polysaccharides or polyhydroxyalkanoates. Additionally, biomass composition is known to shift with growth rate and culturing stress [
45,
81]; the provided approach can be used to create culturing condition-specific cellular growth reactions. Altogether, the current work serves as a useful resource for the broader computational biology community, which will enable more accurate representations of biomass synthesis and therefore more accurate metabolism simulations.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}