Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment



Abstract
:1. Introduction
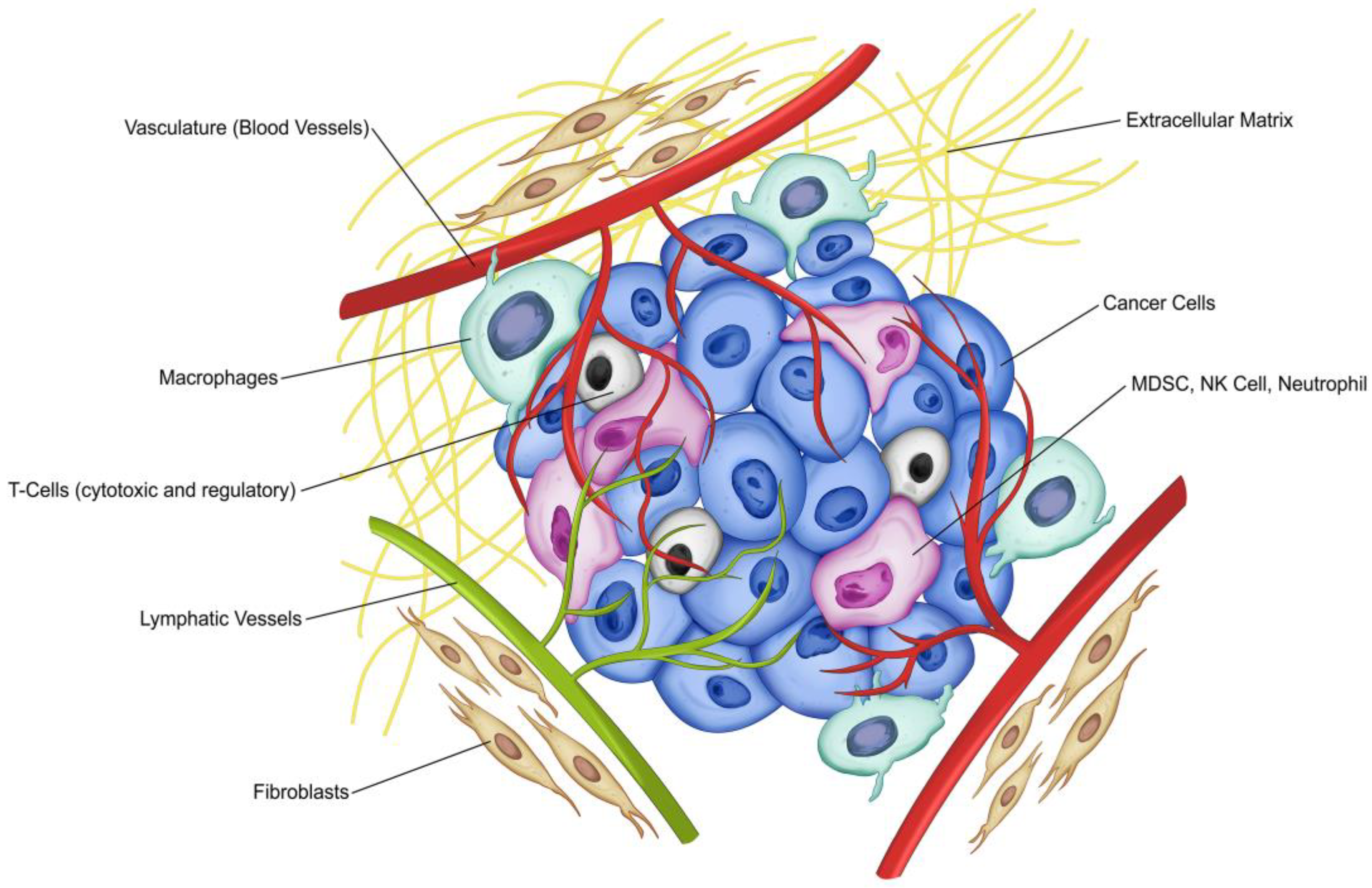
2. Immune System Biology and Cancer
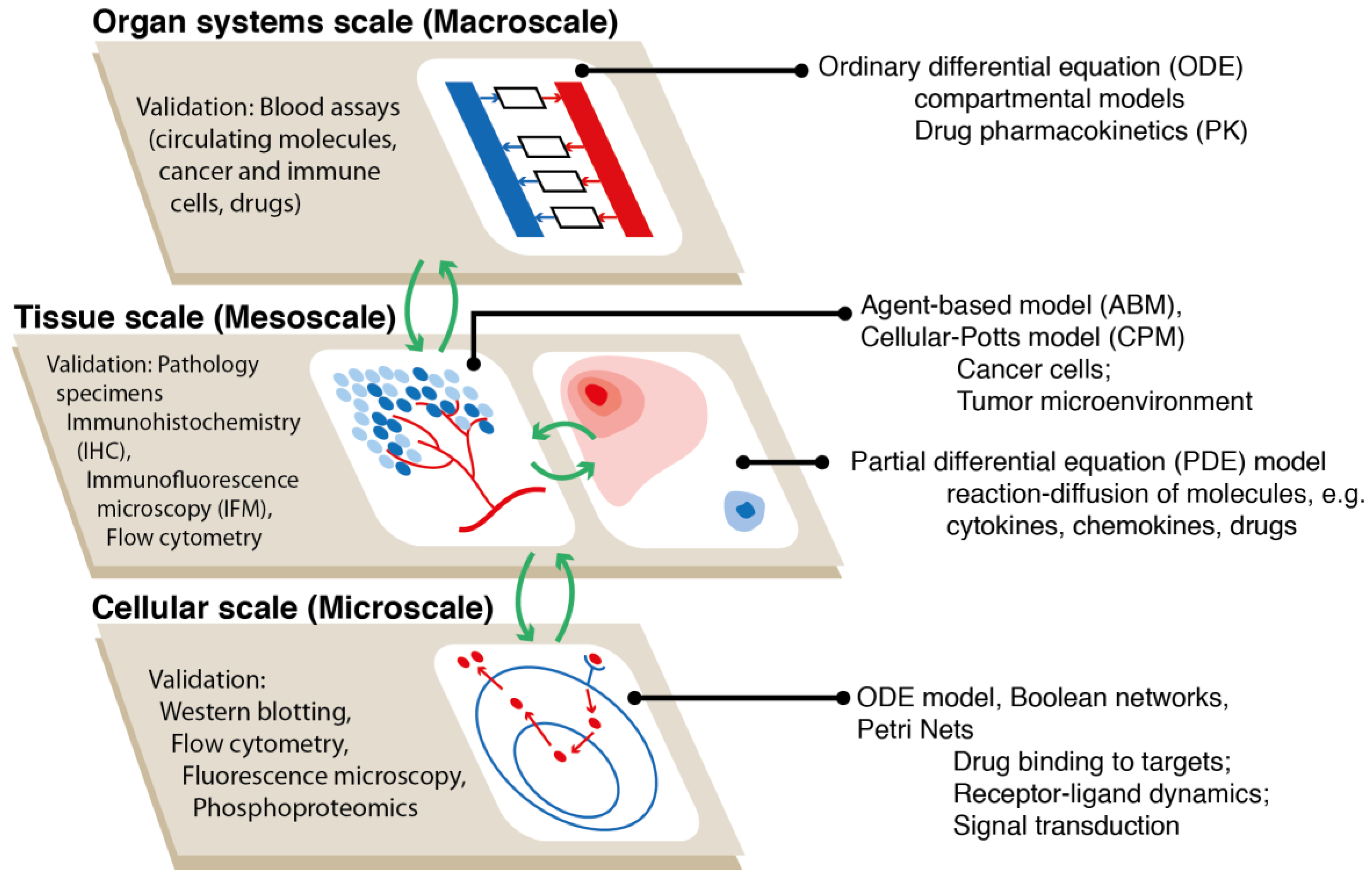
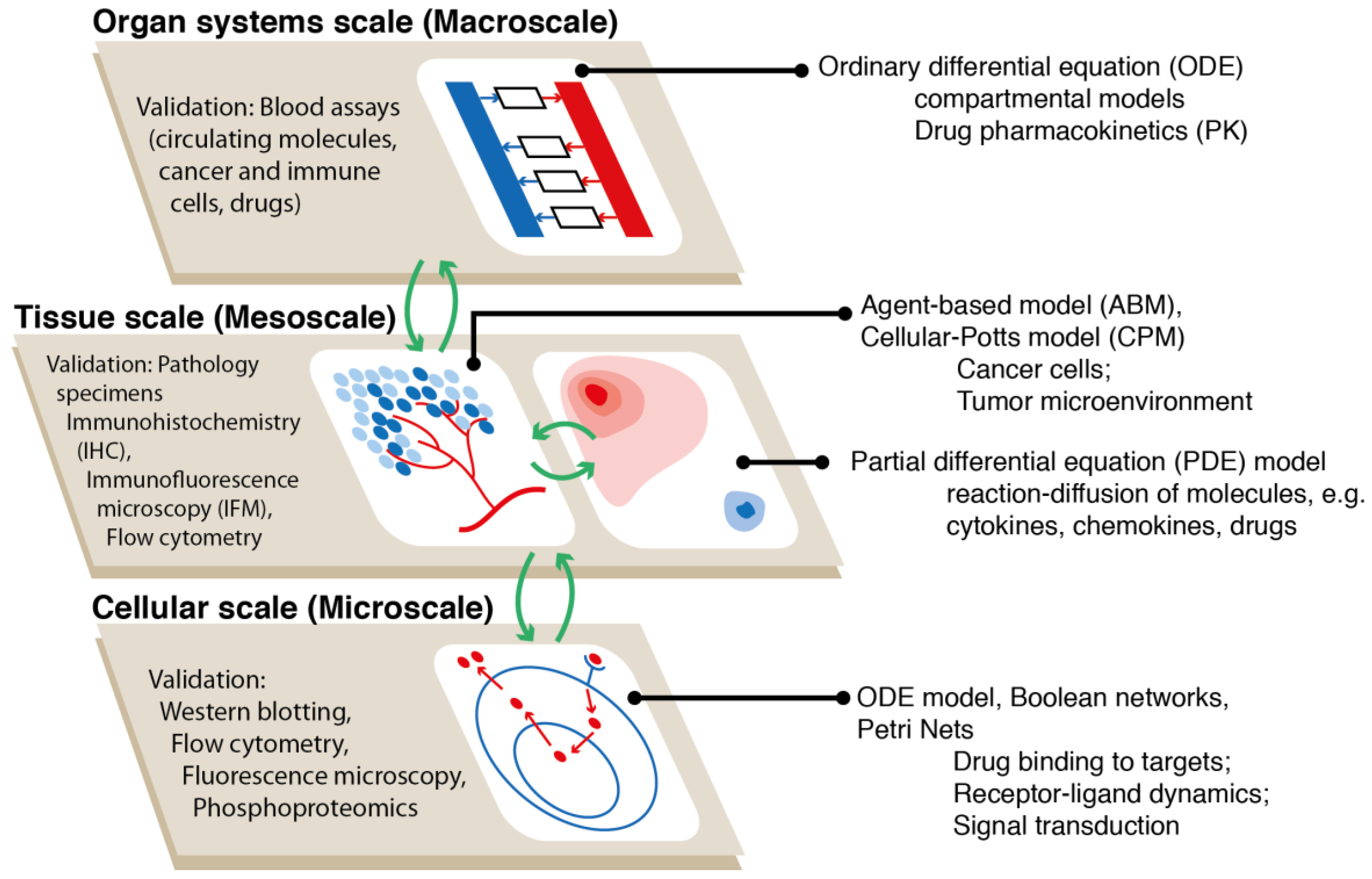
3. Overview of Computational Modeling Methodologies including Agent-Based Modeling
- (1)
- Models focusing on immune-related tumor mechanobiology
- (2)
- Models focusing on tumor-associated vasculature in the immune response
- (3)
- Models focusing on tumor-associated lymphatics and lymph nodes
- (4)
- Models focusing on tumor immunotherapy
- (5)
- Models focusing on tumor-enhancing immune cells
- (6)
- Models focusing on intra-tumor heterogeneity
3.1. Models Focusing on Tumor Mechanobiology
3.2. Models Focusing on Tumor-Associated Vasculature in the Immune Response
3.3. Models Focusing on Tumor-Associated Lymphatics and Lymph Nodes
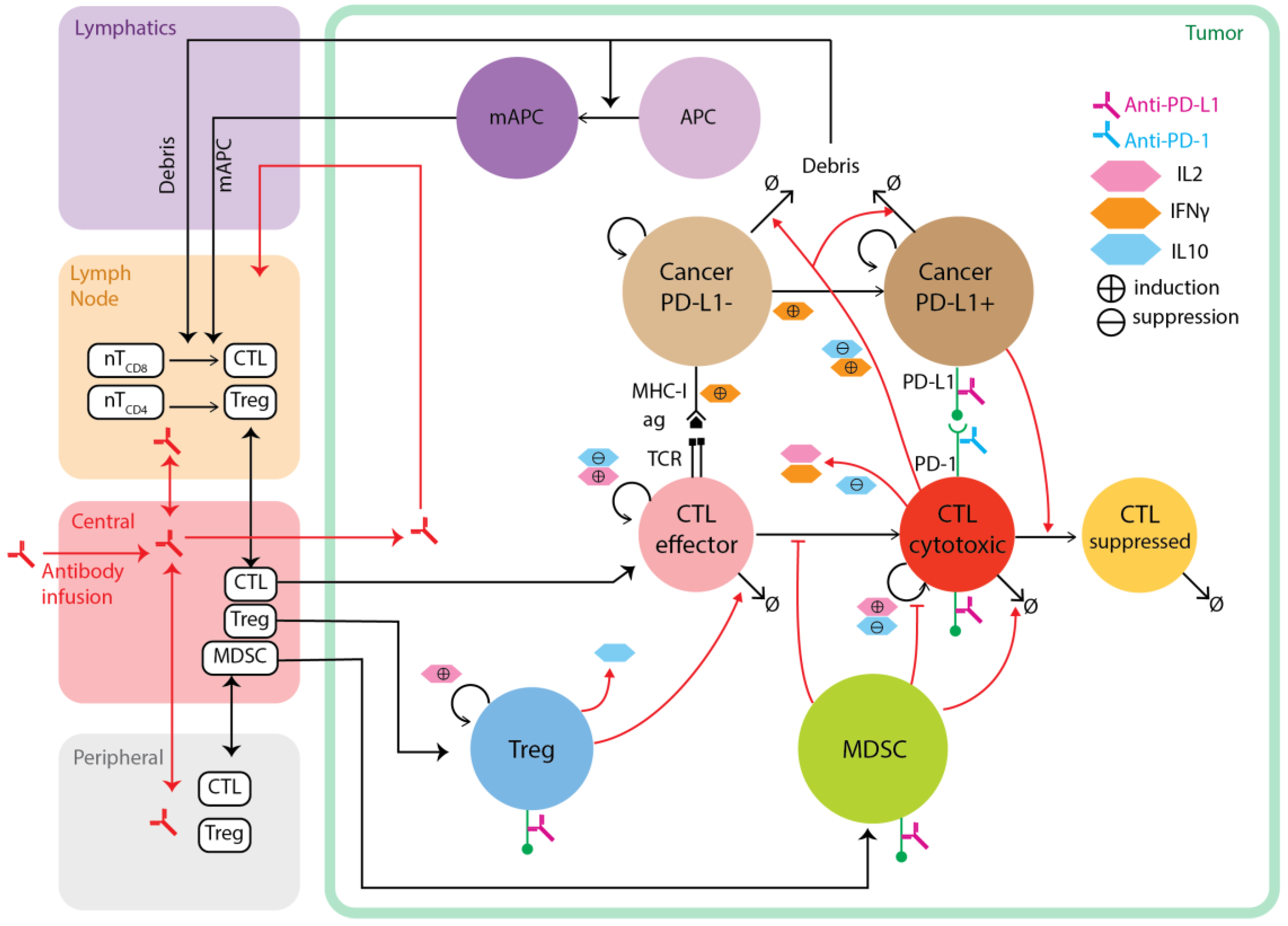
3.4. Models Focusing on Tumor Immunotherapy
3.5. Models Focusing on Tumor-Enhancing Immune Cells
3.6. Models Focusing on Intra-Tumor Heterogeneity
4. Discussion and Emerging Applications
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-Immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Vonderheide, R.H. Cellular composition of the tumor microenvironment. Am. Soc. Clin. Oncol. Educ. B 2013, 33, e91–e97. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Crespo, I.; Coukos, G.; Doucey, M.; Xenarios, I. Modelling approaches to discovery in the tumor microenvironment. J. Cancer Immunol. Ther. 2018, 1, 23–37. [Google Scholar]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; Van Der Meer, J.W.M. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Dykes, S.S.; Hughes, V.S.; Wiggins, J.M.; Fasanya, H.O.; Tanaka, M.; Siemann, D. Stromal cells in breast cancer as a potential therapeutic target. Oncotarget 2018, 9, 23761–23779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, B.; Elkord, E. Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Beyer, M.; Schultze, J.L. Regulatory T cells in cancer. Blood 2006, 108, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Seifert, A.M.; Zeng, S.; Zhang, J.Q.; Kim, T.S.; Cohen, N.A.; Beckman, M.J.; Medina, B.D.; Maltbaek, J.H.; Loo, J.K.; Crawley, M.H.; et al. PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of Imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 2017, 23, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Oyer, J.L.; Gitto, S.B.; Altomare, D.A.; Copik, A.J. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology 2018, 7, e1509819. [Google Scholar] [PubMed]
- Abdel-Wahab, N.; Shah, M.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E. Use of immune checkpoint inhibitors in the treatment of patients with cancer and preexisting autoimmune disease: A systematic review. Ann. Intern. Med. 2018, 168, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of human macrophage M1-M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [PubMed]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M.; Noy, R.; Pollard, J.W.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage—Cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.-L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell 2018, 175, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Burger, G.A.; Danen, E.H.J.; Beltman, J.B. Deciphering epithelial—Mesenchymal transition regulatory networks in cancer through computational approaches. Front. Oncol. 2017, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef]
- Lohela, M.; Casbon, A.-J.; Olow, A.; Bonham, L.; Branstetter, D.; Weng, N.; Smith, J.; Werb, Z. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E5086–E5095. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Cieslewicz, M.; Schellinger, J.G.; Pun, S.H. Synthesis and evaluation of multivalent M2pep peptides for targeting alternatively activated M2 macrophages. J. Control. Release 2016, 224, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef]
- Davis, B.P.; Rothenberg, M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hämmerling, G.J.; Carretero, R.; Beckhove, P.; Salgado, O.C.; Sektioglu, I.M.; Garbi, N. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar]
- Sakkal, S.; Miller, S.; Apostolopoulos, V.; Nurgali, K. Eosinophils in cancer: Favourable or unfavourable? Curr. Med. Chem. 2016, 23, 650–666. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.B.; Kurhekar, M.P. Review of the systems biology of the immune system using agent-based models. IET Syst. Biol. 2018, 12, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A.; McCawley, L.J. Current trends in mathematical modeling of tumor-microenvironment interactions: A survey of tools and applications. Exp. Biol. Med. 2010, 235, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Eftimie, R.; Bramson, J.L.; Earn, D.J.D. Interactions between the immune system and cancer: A brief review of non-spatial mathematical models. Bull. Math. Biol. 2011, 73, 2–32. [Google Scholar] [CrossRef]
- Alemani, D.; Pappalardo, F.; Pennisi, M.; Motta, S.; Brusic, V. Combining cellular automata and lattice Boltzmann method to model multiscale avascular tumor growth coupled with nutrient diffusion and immune competition. J. Immunol. Methods 2012, 376, 55–68. [Google Scholar] [CrossRef]
- Bellomo, N.; Delitala, M. From the mathematical kinetic, and stochastic game theory to modelling mutations, onset, progression and immune competition of cancer cells. Phys. Life Rev. 2008, 5, 183–206. [Google Scholar] [CrossRef]
- Eladdadi, A.; de Pillis, L.; Kim, P. Modelling tumour-immune dynamics, disease progression and treatment. Lett. Biomath. 2018, 5, S1–S5. [Google Scholar] [CrossRef]
- Dritschel, H.; Waters, S.L.; Roller, A.; Byrne, H.M. A mathematical model of cytotoxic and helper T cell interactions in a tumour microenvironment. Lett. Biomath. 2018, 5, S36–S68. [Google Scholar] [CrossRef] [Green Version]
- Nikolopoulou, E.; Johnson, L.R.; Harris, D.; Nagy, J.D.; Stites, E.C.; Kuang, Y. Tumour-immune dynamics with an immune checkpoint inhibitor. Lett. Biomath. 2018, 5, S137–S159. [Google Scholar] [CrossRef]
- Salgia, R.; Mambetsariev, I.; Hewelt, B.; Achuthan, S.; Li, H.; Poroyko, V.; Wang, Y.; Sattler, M. Modeling small cell lung cancer (SCLC) biology through deterministic and stochastic mathematical models. Oncotarget 2018, 9, 26226–26242. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Vella, A.T.; Adler, A.J.; Laubenbacher, R.C. Addressing current challenges in cancer immunotherapy with mathematical and computational modelling. J. R. Soc. Interface 2017, 14, 20170150. [Google Scholar] [CrossRef] [PubMed]
- Chiacchio, F.; Pennisi, M.; Russo, G.; Motta, S.; Pappalardo, F. Agent-based modeling of the immune system: NetLogo, a promising framework. BioMed Res. Int. 2014, 907171. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Mi, Q.; Dutta-Moscato, J.; Vodovotz, Y. Agent-based models in translational systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Chavali, A.K.; Gianchandani, E.P.; Tung, K.S.; Lawrence, M.B.; Peirce, S.M.; Papin, J.A. Characterizing emergent properties of immunological systems with multi-cellular rule-based computational modeling. Trends Immunol. 2008, 29, 589–599. [Google Scholar] [CrossRef]
- Shi, Z.Z.; Wu, C.-H.; Ben-Arieh, D. Agent-based model: A surging tool to simulate infectious diseases in the immune system. Open J. Model. Simul. 2014, 02, 12–22. [Google Scholar] [CrossRef]
- Segovia-Juarez, J.L.; Ganguli, S.; Kirschner, D. Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J. Theor. Biol. 2004, 231, 357–376. [Google Scholar] [CrossRef]
- Tokarski, C.; Hummert, S.; Mech, F.; Figge, M.T.; Germerodt, S.; Schroeter, A.; Schuster, S.; Linde, J.; Hu, G. Agent-based modeling approach of immune defense against spores of opportunistic human pathogenic fungi. Front. Microbiol. 2012, 3, 129. [Google Scholar] [CrossRef]
- Dong, X.; Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Agent-based modeling of endotoxin-induced acute inflammatory response in human blood leukocytes. PLoS ONE 2010, 5, e9249. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, A.; Mi, Q.; Tzen, Y.T.; Brienza, D.; Vodovotz, Y. Hybrid equation/agent-based model of ischemia-induced hyperemia and pressure ulcer formation predicts greater propensity to ulcerate in subjects with spinal cord injury. PLoS Comput. Biol. 2013, 9, e1003070. [Google Scholar] [CrossRef] [PubMed]
- Santoni, D.; Pedicini, M.; Castiglione, F. Implementation of a regulatory gene network to simulate the TH1/2 differentiation in an agent-based model of hypersensitivity reactions. Bioinformatics 2008, 24, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, A.M.; Lawrence, M.B.; Shang, H.; Katz, A.J.; Peirce, S.M. Agent-based model of therapeutic adipose-derived stromal cell trafficking during ischemia predicts ability to roll on p-selectin. PLoS Comput. Biol. 2009, 5, e1000294. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.M.; Lysenko, M.; Marino, S.; Kirschner, D.; Souza, R.M.D.; Arbor, A. Data-parallel algorithms for agent-based model simulation of tuberculosis on graphics processing units. In Proceedings of the 2009 Spring Simulation Multiconference, San Diego, CA, USA, 22–27 March 2009. [Google Scholar]
- Song, S.O.; Hogg, J.; Peng, Z.Y.; Parker, R.; Kellum, J.A.; Clermont, G. Ensemble models of neutrophil trafficking in severe sepsis. PLoS Comput. Biol. 2012, 8, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Mi, Q.; Rivière, B.; Clermont, G.; Steed, D.L.; Vodovotz, Y. Agent-based model of inflammation and wound healing: Insights into diabetic foot ulcer pathology and the role of transforming growth factor-β1. Wound Repair Regen. 2007, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, T.; Byrne, H.M.; Maini, P.K. A mathematical model of the effects of hypoxia on the cell-cycle of normal and cancer cells. J. Theor. Biol. 2004, 229, 395–411. [Google Scholar] [CrossRef]
- Hoehme, S.; Bertaux, F.; Weens, W.; Grasl-Kraupp, B.; Hengstler, J.G.; Drasdo, D. Model prediction and validation of an order mechanism controlling the spatiotemporal phenotype of early hepatocellular carcinoma. Bull. Math. Biol. 2018, 80, 1134–1171. [Google Scholar] [CrossRef]
- Bianca, C.; Pennisi, M. The triplex vaccine effects in mammary carcinoma: A nonlinear model in tune with SimTriplex. Nonlinear Anal. Real World Appl. 2012, 13, 1913–1940. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Jing, C.; Ye, G.; Wu, H.; Miao, H.; Wu, Y.; Zhou, X. Multi-scale agent-based modeling on melanoma and its related angiogenesis analysis. Theor. Biol. Med. Model. 2013, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.A.; Tavernar, L.; Leiss, F.; Herpel, E.; et al. In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res. 2017, 77, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Pappalardo, F.; Motta, S. Agent based modeling of lung metastasis-immune system competition. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2009; Volume 5666, pp. 1–3. [Google Scholar]
- Jagiella, N.; Müller, B.; Müller, M.; Vignon-Clementel, I.E.; Drasdo, D. Inferring growth control mechanisms in growing multi-cellular spheroids of NSCLC cells from spatial-temporal image data. PLoS Comput. Biol. 2016, 12, e1004412. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Pappalardo, F.; Palladini, A.; Nicoletti, G.; Nanni, P.; Lollini, P.-L.; Motta, S. Modeling the competition between lung metastases and the immune system using agents. BMC Bioinform. 2010, 11, S13. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Celada, F.; Ruffo, S.; Seiden, P.E. The transition between immune and disease states in a cellular automaton model of clonal immune response. Phys. A Stat. Mech. Its Appl. 1997, 245, 145–163. [Google Scholar] [CrossRef] [Green Version]
- Baldazzi, V.; Castiglione, F.; Bernaschi, M. An enhanced agent based model of the immune system response. Cell Immunol. 2006, 244, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Bernaschi, M.; Castiglione, F. Design and implementation of an immune system simulator. Comput. Biol. Med. 2001, 31, 303–331. [Google Scholar] [CrossRef]
- Celada, F.; Seiden, P.E. A computer model of cellular interactions in the immune system. Immunol. Today 1992, 13, 56–62. [Google Scholar] [CrossRef]
- Emerson, A.; Rossi, E. ImmunoGrid - the virtual human immune system project. Stud. Heal. Technol. Inf. 2007, 126, 87–92. [Google Scholar]
- Halling-Brown, M.; Pappalardo, F.; Rapin, N.; Zhang, P.; Alemani, D.; Emerson, A.; Castiglione, F.; Duroux, P.; Pennisi, M.; Miotto, O.; et al. ImmunoGrid: Towards agent-based simulations of the human immune system at a natural scale. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 2799. [Google Scholar] [CrossRef]
- Perelson, A.S.; Ribeiro, R.M. Modeling the within-host dynamics of HIV infection. BMC Biol. 2013, 11, 96. [Google Scholar] [CrossRef]
- Warrender, C.; Forrest, S.; Koster, F. Modeling intercellular interactions in early Mycobacterium infection. Bull. Math. Biol. 2006, 68, 2233–2261. [Google Scholar] [CrossRef] [PubMed]
- Ghaffarizadeh, A.; Heiland, R.; Friedman, S.H.; Mumenthaler, S.M.; Macklin, P. PhysiCell: An open source physics-based cell simulator for 3-D multicellular systems. PLOS Comput. Biol. 2018, 14, e1005991. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, J.C.L.; Talkenberger, K.; Seifert, M.; Klink, B.; Hawkins-Daarud, A.; Swanson, K.R.; Hatzikirou, H.; Deutsch, A. The biology and mathematical modelling of glioma invasion: A review. J. R. Soc. Interface 2017, 14, 20170490. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.C.; Rockne, R.C.; Hawkins-Daarud, A.; Gallaher, J.; Anderson, A.R.A.; Canoll, P.; Swanson, K.R. Simulating PDGF-driven glioma growth and invasion in an anatomically accurate brain domain. Bull. Math. Biol. 2018, 80, 1292–1309. [Google Scholar] [CrossRef] [PubMed]
- Juliano, J.; Gil, O.; Hawkins-Daarud, A.; Noticewala, S.; Rockne, R.C.; Gallaher, J.; Massey, S.C.; Sims, P.A.; Anderson, A.R.A.; Swanson, K.R.; et al. Comparative dynamics of microglial and glioma cell motility at the infiltrative margin of brain tumours. J. R. Soc. Interface 2018, 15, 20170582. [Google Scholar] [CrossRef] [PubMed]
- Frascoli, F.; Flood, E.; Kim, P.S. A model of the effects of cancer cell motility and cellular adhesion properties on tumour-immune dynamics. Math. Med. Biol. 2016, 34, dqw004. [Google Scholar] [CrossRef]
- Noonan, D.M.; De Lerma Barbaro, A.; Vannini, N.; Mortara, L.; Albini, A. Inflammation, inflammatory cells and angiogenesis: Decisions and indecisions. Cancer Metastasis Rev. 2008, 27, 31–40. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Uppal, A.; Wightman, S.C.; Ganai, S.; Weichselbaum, R.R.; An, G. Investigation of the essential role of platelet-tumor cell interactions in metastasis progression using an agent-based model. Theor. Biol. Med. Model. 2014, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Alfonso, J.C.L.; Schaadt, N.S.; Schönmeyer, R.; Brieu, N.; Forestier, G.; Wemmert, C.; Feuerhake, F.; Hatzikirou, H. In-silico insights on the prognostic potential of immune cell infiltration patterns in the breast lobular epithelium. Sci. Rep. 2016, 6, 33322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.P.; Krouskop, T.A.; Newell, P.H. A computer model of the lymphatic system. Comput. Biol. Med. 1977, 7, 181–197. [Google Scholar] [CrossRef]
- Jamalian, S.; Jafarnejad, M.; Zawieja, S.D.; Bertram, C.D.; Gashev, A.A.; Zawieja, D.C.; Davis, M.J.; Moore, J.E. Demonstration and analysis of the suction effect for pumping lymph from tissue beds at subatmospheric pressure. Sci. Rep. 2017, 7, 12080. [Google Scholar] [CrossRef] [PubMed]
- Jamalian, S.; Davis, M.J.; Zawieja, D.C.; Moore, J.E. Network scale modeling of lymph transport and its effective pumping parameters. PLoS ONE 2016, 11, e0148384. [Google Scholar] [CrossRef] [PubMed]
- Jamalian, S.; Bertram, C.D.; Richardson, W.J.; Moore, J.E. Parameter sensitivity analysis of a lumped-parameter model of a chain of lymphangions in series. Am. J. Physiol. Circ. Physiol. 2013, 305, H1709–H1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.T.; van Loon, R.; Wang, W.; Zawieja, D.C.; Moore, J.E. Determining the combined effect of the lymphatic valve leaflets and sinus on resistance to forward flow. J. Biomech. 2015, 48, 3584–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roose, T.; Swartz, M.A. Multiscale modeling of lymphatic drainage from tissues using homogenization theory. J. Biomech. 2012, 45, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Jafarnejad, M.; Zawieja, D.C.; Brook, B.S.; Nibbs, R.J.B.; Moore, J.E. A novel computational model predicts key regulators of chemokine gradient formation in lymph nodes and site-specific roles for CCL19 and ACKR4. J. Immunol. 2017, 199, ji1700377. [Google Scholar] [CrossRef]
- Jafarnejad, M.; Woodruff, M.C.; Zawieja, D.C.; Carroll, M.C.; Moore, J.E. Modeling lymph flow and fluid exchange with blood vessels in lymph nodes. Lymphat. Res. Biol. 2015, 13, 234–247. [Google Scholar] [CrossRef]
- Cooper, L.J.; Heppell, J.P.; Clough, G.F.; Ganapathisubramani, B.; Roose, T. An image-based model of fluid flow through lymph nodes. Bull. Math. Biol. 2016, 78, 52–71. [Google Scholar] [CrossRef]
- Marino, S.; Gideon, H.P.; Gong, C.; Mankad, S.; McCrone, J.T.; Lin, P.L.; Linderman, J.J.; Flynn, J.A.L.; Kirschner, D.E. Computational and empirical studies predict mycobacterium tuberculosis-specific T cells as a biomarker for infection outcome. PLoS Comput. Biol. 2016, 12, e1004804. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Linderman, J.J.; Kirschner, D. Harnessing the heterogeneity of T cell differentiation fate to fine-tune generation of effector and memory T cells. Front. Immunol. 2014, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Margaris, K.N.; Black, R.A. Modelling the lymphatic system: Challenges and opportunities. J. R. Soc. Interface 2012, 9, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M. A mathematical model for the germinal center morphology and affinity maturation. J. Theor. Biol. 2002, 216, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M.E.; Maini, P.K. Cutting edge: Back to “one-way” germinal centers. J. Immunol. 2005, 174, 2489–2493. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M.E.; Maini, P.K.; Iber, D. An analysis of B cell selection mechanisms in germinal centers. Math. Med. Biol. A J. IMA 2006, 23, 255–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogle, G.; Dunbar, P.R. T cell responses in lymph nodes. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 107–116. [Google Scholar] [CrossRef]
- Bogle, G.; Dunbar, P.R. Agent-based simulation of T-cell activation and proliferation within a lymph node. Immunol. Cell Biol. 2010, 88, 172–179. [Google Scholar] [CrossRef]
- Bogle, G.; Dunbar, P.R. On-lattice simulation of T cell motility, chemotaxis, and trafficking in the lymph node paracortex. PLoS ONE 2012, 7, e45258. [Google Scholar] [CrossRef]
- Bogle, G.; Dunbar, P.R. Simulating T-cell motility in the lymph node paracortex with a packed lattice geometry. Immunol. Cell Biol. 2008, 86, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Moreau, H.D.; Bogle, G.; Bousso, P. A virtual lymph node model to dissect the requirements for T-cell activation by synapses and kinapses. Immunol. Cell Biol. 2016, 94, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folcik, V.A.; An, G.C.; Orosz, C.G. The Basic Immune Simulator: An agent-based model to study the interactions between innate and adaptive immunity. Theor. Biol. Med. Model. 2007, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.S.; Levy, D.; Lee, P.P. Modeling and simulation of the immune system as a self-regulating network. Methods Enzymol. 2009, 467, 79–109. [Google Scholar] [PubMed]
- Jacob, C.; Sarpe, V.; Gingras, C.; Feyt, R.P. Swarm-based simulations for immunobiology: What can agent-based models teach us about the immune system? In Intelligent Systems Reference Library; Springer: Berlin/Heidelberg, Germany, 2011; Volume 11, pp. 29–64. [Google Scholar]
- Marino, S.; El-Kebir, M.; Kirschner, D. A hybrid multi-compartment model of granuloma formation and T cell priming in Tuberculosis. J. Theor. Biol. 2011, 280, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, S.; Kirschner, D. A multi-compartment hybrid computational model predicts key roles for dendritic cells in Tuberculosis infection. Computation 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Dréau, D.; Stanimirov, D.; Carmichael, T.; Hadzikadic, M. An agent-based model of solid tumor progression. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2009; Volume 5462, pp. 187–198. [Google Scholar]
- Pappalardo, F.; Forero, I.M.; Pennisi, M.; Palazon, A.; Melero, I.; Motta, S. Simb16: Modeling induced immune system response against B16-melanoma. PLoS ONE 2011, 6, e26523. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolaños, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Santiago, D.N.; Heidbuechel, J.P.W.; Kandell, W.M.; Walker, R.; Djeu, J.; Engeland, C.E.; Abate-Daga, D.; Enderling, H. Fighting cancer with mathematics and viruses. Viruses 2017, 9, 239. [Google Scholar] [CrossRef]
- Walker, R.; Navas, P.E.; Friedman, S.H.; Galliani, S.; Karolak, A.; MacFarlane, F.; Noble, R.; Poleszczuk, J.; Russell, S.; Rejniak, K.A.; et al. Enhancing synergy of CAR T cell therapy and oncolytic virus therapy for pancreatic cancer. bioRxiv 2016, 055988. [Google Scholar] [CrossRef]
- Rohrs, J.A.; Zheng, D.; Graham, N.A.; Wang, P.; Finley, S.D. Computational model of chimeric antigen receptors explains site-specific phosphorylation kinetics. Biophys. J. 2018, 115, 1116–1129. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Milberg, O.; Wang, B.; Vicini, P.; Narwal, R.; Roskos, L.; Popel, A.S. A computational multiscale agent-based model for simulating spatio-temporal tumour immune response to PD1 and PDL1 inhibition. J. R. Soc. Interface 2017, 14, 20170320. [Google Scholar] [CrossRef] [PubMed]
- Hillen, T.; Enderling, H.; Hahnfeldt, P. The tumor growth paradox and immune system-mediated selection for cancer stem cells. Bull. Math. Biol. 2013, 75, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Enderling, H.; Hlatky, L.; Hahnfeldt, P. Immunoediting: Evidence of the multifaceted role of the immune system in self-metastatic tumor growth. Theor. Biol. Med. Model. 2012, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.K.; Chuang, Y.; Knapp, L.M.; Brockmann, D.; Kath, W.L.; Leonard, J.N. Spatial and functional heterogeneities shape collective behavior of tumor-immune networks. PLoS Comput. Biol. 2015, 11, e1004181. [Google Scholar] [CrossRef]
- Knútsdóttir, H.; Pálsson, E.; Edelstein-Keshet, L. Mathematical model of macrophage-facilitated breast cancer cells invasion. J. Theor. Biol. 2014, 357, 184–199. [Google Scholar] [CrossRef]
- Knutsdottir, H.; Condeelis, J.S.; Palsson, E. 3-D individual cell based computational modeling of tumor cell-macrophage paracrine signaling mediated by EGF and CSF-1 gradients. Integr. Biol. 2016, 8, 104–119. [Google Scholar] [CrossRef]
- Norton, K.A.; Jin, K.; Popel, A.S. Modeling triple-negative breast cancer heterogeneity: Effects of stromal macrophages, fibroblasts and tumor vasculature. J. Theor. Biol. 2018, 452, 56–68. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Lehrach, H. Virtual clinical trials, an essential step in increasing the effectiveness of the drug development process. Public Health Genom. 2015, 18, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Pourhasanzade, F.; Sabzpoushan, S.; Alizadeh, A.M.; Esmati, E. An agent-based model of avascular tumor growth: Immune response tendency to prevent cancer development. Simulation 2017, 93, 641–657. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Bucci, V.; Akkari, L.; Deforet, M.; Joyce, J.A.; Xavier, J.B. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc. Natl. Acad. Sci. USA 2013, 110, 19402–19407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueredo, G.P.; Aickelin, U.; Siebers, P.O. Systems dynamics or agent-based modelling for immune simulation? In Proceedings of the Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2011; Volume 6825, pp. 81–94. [Google Scholar]
- Figueredo, G.P.; Siebers, P.-O.; Aickelin, U. Investigating mathematical models of immuno-interactions with early-stage cancer under an agent-based modelling perspective. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef]
- Figueredo, G.P.; Joshi, T.V.; Osborne, J.M.; Byrne, H.M.; Owen, M.R. On-lattice agent-based simulation of populations of cells within the open-source Chaste framework. Interface Focus 2013, 3, 20120081. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, G.P.; Siebers, P.O.; Owen, M.R.; Reps, J.; Aickelin, U. Comparing stochastic differential equations and agent-based modelling and simulation for early-stage cancer. PLoS ONE 2014, 9, e95150. [Google Scholar] [CrossRef] [PubMed]
- Yankeelov, T.E.; An, G.; Saut, O.; Luebeck, E.G.; Popel, A.S.; Ribba, B.; Vicini, P.; Zhou, X.; Weis, J.A.; Ye, K.; et al. Multi-scale modeling in clinical oncology: Opportunities and barriers to success. Ann. Biomed. Eng. 2016, 44, 2626–2641. [Google Scholar] [CrossRef]
- Cosgrove, J.; Butler, J.; Alden, K.; Read, M.; Kumar, V.; Cucurull-Sanchez, L.; Timmis, J.; Coles, M. Agent-based modeling in systems pharmacology. CPT Pharmacometrics Syst. Pharmacol. 2015, 4, 615–629. [Google Scholar] [CrossRef]
- Rieger, T.R.; Allen, R.J.; Bystricky, L.; Chen, Y.; Colopy, G.W.; Cui, Y.; Gonzalez, A.; Liu, Y.; White, R.D.; Everett, R.A.; et al. Improving the generation and selection of virtual populations in quantitative systems pharmacology models. Prog. Biophys. Mol. Biol. 2018, 139, 15–22. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolín, A.A.; Deu-Pons, J.; Perez-Llamas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 2015, 27, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A.; Maini, P.K. Mathematical oncology. Bull. Math. Biol. 2018, 80, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, B. Mathematical modeling and computational prediction of cancer drug resistance. Brief. Bioinform. 2017, 19, 1382–1399. [Google Scholar] [CrossRef]
- Arney, K. Improving brain-cancer therapies through mathematical modelling. Nature 2018, 561, S52–S53. [Google Scholar] [CrossRef] [PubMed]
- Barua, D.; Hlavacek, W.S. Modeling the effect of APC truncation on destruction complex function in colorectal cancer cells. PLoS Comput. Biol. 2013, 9, e1003217. [Google Scholar] [CrossRef]
- Klinger, B.; Sieber, A.; Fritsche-Guenther, R.; Witzel, F.; Berry, L.; Schumacher, D.; Yan, Y.; Durek, P.; Merchant, M.; Schafer, R.; et al. Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol. Syst. Biol. 2014, 9, 673. [Google Scholar] [CrossRef]
- Kirouac, D.C.; Du, J.Y.; Lahdenranta, J.; Overland, R.; Yarar, D.; Paragas, V.; Pace, E.; McDonagh, C.F.; Nielsen, U.B.; Onsum, M.D. Computational modeling of ERBB2-amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors (Science Signaling 6:288 (ra68)). Sci. Signal. 2014, 7, er5. [Google Scholar]
- Carbo, A.; Bassaganya-Riera, J.; Pedragosa, M.; Viladomiu, M.; Marathe, M.; Eubank, S.; Wendelsdorf, K.; Bisset, K.; Hoops, S.; Deng, X.; et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS ONE 2013, 8, e73365. [Google Scholar] [CrossRef]
- Zand, R.; Abedi, V.; Hontecillas, R.; Lu, P.; Noorbakhsh-Sabet, N.; Verma, M.; Leber, A.; Tubau-Juni, N.; Bassaganya-Riera, J. Development of synthetic patient populations and in silico clinical trials. In Accelerated Path to Cures; Springer International Publishing AG: Basel, Switzerland, 2018; pp. 57–77. [Google Scholar]
- Edelman, L.B.; Eddy, J.A.; Price, N.D. In silico models of cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 438–459. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Anders, R.A.; Zhu, Q.; Taube, J.M.; Green, B.; Cheng, W.; Bartelink, I.H.; Vicini, P.; Wang, B.; Popel, A.S. Quantitative Characterization of CD8+ T Cell Clustering and Spatial Heterogeneity in Solid Tumors. Front Oncol. 2018, 8, 649. [Google Scholar] [CrossRef]
- D’Esposito, A.; Sweeney, P.W.; Ali, M.; Saleh, M.; Ramasawmy, R.; Roberts, T.A.; Agliardi, G.; Desjardins, A.; Lythgoe, M.F.; Pedley, R.B.; et al. Computational fluid dynamics with imaging of cleared tissue and of in vivo perfusion predicts drug uptake and treatment responses in tumours. Nat. Biomed. Eng. 2018, 2, 773–787. [Google Scholar] [CrossRef]
- Stamatelos, S.K.; Kim, E.; Pathak, A.P.; Popel, A.S. A bioimage informatics based reconstruction of breast tumor microvasculature with computational blood flow predictions. Microvasc. Res. 2014, 91, 8–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirouac, D.C. How do we “validate” a QSP model? CPT Pharmacometrics Syst. Pharmacol. 2018, 7, 547–548. [Google Scholar] [CrossRef]
- Gadkar, K.; Kirouac, D.; Parrott, N.; Ramanujan, S. Quantitative systems pharmacology: A promising approach for translational pharmacology. Drug Discov. Today Technol. 2016, 21–22, 57–65. [Google Scholar] [CrossRef]
- Workman, P.; Draetta, G.F.; Schellens, J.H.M.; Bernards, R. How much longer will we put up with 100,000 cancer drugs? Cell 2017, 168, 579–583. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Pan, X.; Xiaoqiang, L.; Mohan, R.; Komaki, R.; Cox, J.D.; Chang, J.Y. Intensity-modulated proton therapy reduces the dose to normal tissue compared with intensity-modulated radiation therapy or passive scattering proton therapy and enables individualized radical radiotherapy for extensive stage IIIB non-small-cell lung canc. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 357–366. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| 3.1. Models Focusing on Immune-Related Tumor Mechanobiology | 3.2 Models Focusing on Tumor-Associated Vasculature in the Immune Response | 3.3 Models Focusing on Tumor-Associated Lymphatics | 3.4 Models Focusing on Tumor Immunotherapy | 3.5 Models Focusing on Tumor-Enhancing Immune Cells | 3.6 Models Focusing on Intra-Tumor Heterogeneity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | Ref | Study | Ref | Study | Ref | Study | Ref | Study | Ref | Study | Ref |
| Cellular adhesion to ECM | (Frascoli et al. 2016) [89] | Early metastasis | (Uppal et al. 2017) [92] | Germinal centers of LN | (Meyer-Hermann et al. 2002, 2005) [106,107,108] | Lung met in mammary carcinoma | (Pennisi et al. 2009) [73] | Cancer stem cell-immune cell interaction | (Hillen 2013) (Enderling 2012) [128,129] | Tumor, NK cell, cytotoxic T-cell interactions | (Pourhasanzade et al. 2017) [137] |
| Adoptive cell transfer in colorectal cancer | (Kather et al. 2017) [72] | Immune-epithelial cell interactions in breast epithelium | (Alfonso et al. 2016) [93] | T-cell behavior in LN | (Bogle et al. 2010, 2012, 2008) [109,110,111,112,113] | Effect of vaccine on lung metastasis | (Pennisi et al. 2010) [75] | Effect of M1 and M2 macrophages on tumor growth | (Wells 2015) [131] | Effect of stroma on tumor spatial patterns | (Carmona-Fontaine et al. 2013) [138] |
| T-cell activation in virtual LN | (Moreau, 2016) [113] | Immunotherapy in solid tumors | (Dréau et al. 2009) [119] | Signaling between macrophages and cancer cells | (Knútsdóttir et al. 2014, 2016) [132,133] | Immune cell, macrophage, tumor cell interactions | (Figueredo 2011, 2013) [139,140] | ||||
| Model of LN to study cancer vaccines | (Kim et al. 2009) [100] | Role of T-cells in response to immunotherapy | (Pappalardo et al. 2011) [120] | Effect of macrophages on TNBC tumor growth | (Norton et al. 2018) [134] | Tumors under oxygen-dependent proliferation | (Figueredo 2013, 2014) [141,142] | ||||
| Immune response against viruses | (Jacob et al. 2011) [116] | Effect of different therapies on pancreatic tumors | (Walker et al. 2016) [123] | ||||||||
| Recruitment of APCs in the LN from lung | (Marino et al. 2011) [117] | Spatio-temporal dynamics of tumor-immune cell interactions | (Gong et al. 2017) [127] | ||||||||
| T-cell trafficking and proliferation | (Marino et al. 2016) [118] | ||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norton, K.-A.; Gong, C.; Jamalian, S.; Popel, A.S. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. https://doi.org/10.3390/pr7010037
Norton K-A, Gong C, Jamalian S, Popel AS. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes. 2019; 7(1):37. https://doi.org/10.3390/pr7010037
Chicago/Turabian StyleNorton, Kerri-Ann, Chang Gong, Samira Jamalian, and Aleksander S. Popel. 2019. "Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment" Processes 7, no. 1: 37. https://doi.org/10.3390/pr7010037
APA StyleNorton, K.-A., Gong, C., Jamalian, S., & Popel, A. S. (2019). Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes, 7(1), 37. https://doi.org/10.3390/pr7010037