Abstract
Over the last two decades, understanding of the attachment of colloids to fluid interfaces has attracted the interest of researchers from different fields. This is explained by considering the ubiquity of colloidal and interfacial systems in nature and technology. However, to date, the control and tuning of the assembly of colloids at fluid interfaces remain a challenge. This review discusses some of the most fundamental aspects governing the organization of colloidal objects at fluid interfaces, paying special attention to spherical particles. This requires a description of different physicochemical aspects, from the driving force involved in the assembly to its thermodynamic description, and from the interactions involved in the assembly to the dynamics and rheological behavior of particle-laden interfaces.
1. Introduction
Many dietary products, such as mayonnaise or milk, are good examples of emulsions, which consist of dispersions of liquid droplets in a continuous phase formed by an immiscible liquid. The adsorption of surface-active molecules at the droplet-continuous phase interface is essential for the stabilization of these systems. Similarly, the stability of foams appearing in some beverages, such as beer, is crucial to preventing the fast evaporation of carbon dioxide from the beer in the glass. In general, liquid foams consist of a dispersion of air bubbles in a continuous aqueous phase [1,2,3,4]. Indeed, their stability is only possible as a result of the adsorption of surface-active molecules at the bubble-aqueous phase. Foams and emulsions are also present in pharmaceutical formulations, as well as in shampoos and bath creams, affecting the sensorial perception associated with the use of the products (softness, creaminess, etc.), as well as the feeling of cleanliness [5,6,7,8].
Considering the above examples, it is easily to conclude that the adsorption and assembly of surface-active molecules (e.g., surfactant, (bio)polymers, and micro- and nanoparticles) at fluid interfaces provide the basic building blocks for the creation of emulsions and foams [9], remarkably important in different areas with scientific and technological impacts [10,11,12,13,14]. In particular, the interaction of colloidal particles with interfaces plays a central role in many processes with an industrial interest, including metal recovery by flotation, tertiary oil recovery, interfacial biocatalysis, gas storage, and biomass conversion [14,15,16,17]. Furthermore, biomedical processes, such as the inhalation and transport of colloidal particles through the respiratory tract, involve the interactions of colloidal systems and fluid interfaces [18,19].
In addition, the symmetry breaking and the dimensional confinement appearing from the presence of an interface provides a versatile platform for designing and manufacturing nanomaterials (e.g., materials based in quasi-two-dimensional (quasi-2d) colloidal arrays, where the interfaces are templates or scaffolds for the assembly of colloidal systems). The combination of this geometrical restriction with the prospect of tuning and controlling the assembly of colloidal objects has very recently opened a new avenue for building new functional materials with potential applications for chemical synthesis, separation of chemical compounds, and reconfigurable devices.
On the basis of the above discussion, the understanding of the most fundamental aspects involved in the fabrication and control of colloids assembled at fluid interfaces is of paramount importance to improving their use in the design of new technological systems [20,21,22]. Future developments of the colloidal confinement could enable fabrication of biomimetic and active materials, which could respond to an external perturbation, and even alter their environment. This requires an understanding of the physicochemical bases underlying the behavior of such complex interfaces, which requires a deeper understanding of the physicochemical bases governing the assembly of quasi-2D materials, as well as on the dynamic and equilibrium properties of the assembled systems [3,19,23,24,25,26,27,28,29,30,31,32,33,34,35,36].
In summary, this review presents a general perspective of the most fundamental aspects related to the attachment of colloids at the fluid interface, and the physicochemical properties of the assembled systems, paying special attention to the understanding of the behavior of spherical particles confined at fluid interfaces. In addition, some specific aspect related to the behavior of other colloidal systems at fluid interfaces are briefly discussed.
2. Driving the Assembly: Interfacial Tension and Contact Angle
2.1. Interfacial Tension
The driving force responsible for the adsorption of molecules at a fluid interface is associated with a decrease of the interfacial tension, and therefore the free energy associated with the formation of the interface [7,8,10,11,12,13]. Therefore, the adsorption of amphiphilic molecules (e.g., from surfactants to (bio)polymers) and colloidal particles (including the ones with two regions presenting a different chemical nature, the so-called Janus particles [37,38,39]) at fluid interfaces is driven by the minimization of the Gibbs free energy owing to a decrease of the contact area between the two fluids. This can be explained on the basis of a decrease of the interfacial tension, γ, which may be defined as (i) the force per unit of length acting tangentially at the interface along the contact line between the two phases, or (ii) the energy cost, Gγ, associated with the generation of one unit of contact area, A, between two immiscible fluids at constant values of pressure (p) and temperature (T), i.e., .
It is worth mentioning that the adsorption of colloidal particles at fluid interfaces does not lead to a decrease of the interfacial tension between the two fluid phases, , at the microscopic level as occurs when traditional surfactants are involved. However, a change of this property at the macroscopic level is generally found. This decrease can be understood considering the appearance of a 2D lateral pressure (Π) within the interface due to the entrapment of particles. This lateral pressure is opposed to the tendency of the interface to contract and minimize its area. Therefore, it is possible to assume that the increase of the interfacial packing leads to a measurable decrease of the interfacial tension [40,41,42,43,44,45,46,47,48,49]. Thus, the different origins found for the surface tensions of conventional surfactants and particles at fluid interfaces have limited the development of accurate thermodynamics models describing the behavior of particle-laden interfaces.
The application of conventional state equations (for example, Langmuir, Frumkin, etc. adsorption isotherms) for a thermodynamics description of the interfaces coated by amphiphiles is a mature field of research [50,51]. However, the description of the behavior of colloidal particles trapped at fluid interfaces has been more elusive, mainly due to the differences existing between the sizes of the colloids and the solvent molecules. This makes it necessary to develop a new thermodynamics framework which enables a correct description of particle-laden interfaces. There have been several attempts to provide an accurate thermodynamics description of particle-laden interfaces. The first one was provided by Binks [4] combining the Volmer equation, which accounts for the absence of lateral interactions between particles at low packing density, with the van der Waals equation, which enables describing the lateral interactions between the particles occurring at high packing density. This model was based on the following two main assumptions: (i) each particle behaves as a surfactant molecule and (ii) the area of each particle is finite and similar to its geometrical area. However, this description failed because it only provided a correct prediction for the surface tension at high interfacial densities when the role of the interparticle interactions was important. Experimental findings showed that even at long interparticle distances the interactions play a central role for controlling the interfacial properties which differs from the behavior found for surfactant monolayers. Thus, the differences between particles and surfactant molecules, and the role of the interparticle interactions resulted in failure of the model.
An improved description was provided by Miller’s group [52,53]. They developed a model based on those previously used for describing the adsorption of proteins at fluid interfaces [54], including specific features to account for the behavior of the particle-laden interfaces. Thus, they defined the surface pressure ( = , with and being the surfaces tension for the bare interface and the interface after the adsorption of colloidal particles, respectively) for a particle-laden interface as follows:
where provides information related to the interfacial coverage and gives information of the particle area, with being the so-called cohesion pressure, which gives information about the interactions occurring along the interface, and kB and T the Boltzmann constant and the absolute temperature, respectively. This model provides a good description of the behavior of particles trapped at a fluid interface far from a close-packed state, independently of the size of the particles and their chemical nature.
An alternative description of the thermodynamic behavior of particles at fluid interfaces was proposed by Groot and Stoyanov [55]. This considered a dependence of the interactions with the interfacial packing, resulting in the following expression:
where d is the distance within which the long-range interactions occurs, and Z is the compressibility factor [56]. and y represent the effective diameter of the particles and the interfacial coverage, respectively, with b and b2 being interactions parameters related to the interaction potential. This model was successfully tested by Deshmukh et al. [57]. The development of more refined models describing the thermodynamics behavior of particle-laden interfaces requires a detailed consideration of the interparticle interactions, as well as the wettability of the particles. Furthermore, the inclusion of morphological and structural aspects is expected to help with an accurate description.
2.2. Contact Angle and Wetting
The energy associated with the entrapment of colloidal particles at fluid interfaces (trapping energy ∆Ep) frequently exceeds the thermal energy, kBT, which is the opposite to what usually happens when surfactant molecules are concerned [58]. Thus, assuming the simplest case, the adsorption of a chemically homogeneous spherical particle (without any significant roughness) at an interface between two immiscible fluids, it is possible to define the energy for trapping a particle at the interface, ∆Ep, as the difference between the energy of the particle dispersed in one of the phases, and the energy of the same particle at the fluid interface.
where R is the radius of the particle and indicates the interfacial tension between the two fluid phases. The sign ± is associated with the relative position of the particles with respect to the interface, with + and − signs giving an indication that particles are placed above (hydrophobic particles) or below (hydrophilic particles) the interfacial plane. Equation (3) introduces the parameter θ, which is the contact angle of the colloidal particles attached at the fluid interface, giving a measurement of the particle wettability for each phase, according to Figure 1. This is clear considering an interface between water and an apolar fluid, in which particles with θ < 90° are assumed to be hydrophilic, whereas those with θ > 90° are defined as hydrophobic. It is worth mentioning that the preferential partition of particles between the two phases plays a central role in the relaxation mechanisms appearing in particle-laden interfaces, and as matter of fact in their potential technological applications [59,60].
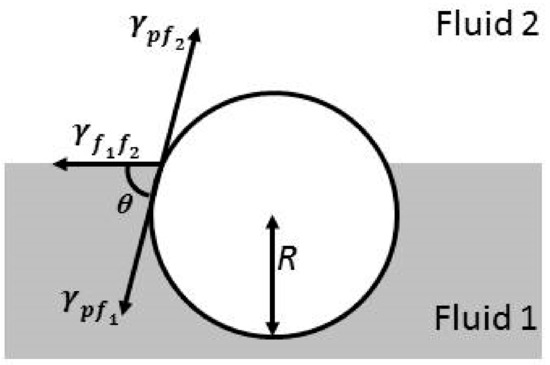
Figure 1.
Scheme of a typical situation for the trapping of a particle at an arbitrary fluid interface, where θ represents the contact angle or relative wettability of the particle by the interface, R is the particle radius, and , , and are the interfacial tensions corresponding to the fluid-fluid interface and the two fluid-solid (and fluid-particle) interfaces, respectively.
On the basis of the scheme shown in Figure 1, and assuming that Young’s law is fulfilled, Equation (4), i.e., there is mechanical equilibrium, it is possible to define the contact angle
where and represent the contact angles of the particles with the polar and apolar phases, respectively; and , y, are referred to the interfacial tensions corresponding to the two fluid-solid interfaces. The relative position of a colloidal particle with respect to the interfacial plane is always defined by its contact angle. Equation (6) is a generalization giving a description of the attachment of spherical particles at the interface between two fluids. However, it is possible to make different modifications on such an equation to include the role of particle geometry and roughness, its surface chemical heterogeneity, and even the effect of the line tension, particularly when nanoparticles are considered [31,61,62]. Thus, Equation (6) is considered as a simple model enabling the evaluation of the assembly process of colloidal objects at fluid interfaces, depending on the following three experimentally accessible parameters: contact angle, interfacial tension between the fluid phases, and particle dimensions.
On the basis of Equation (5), it is possible to assume that microparticles adsorb at fluid interfaces irreversibly, with entrapment energy exceeding many times the thermal energy. However, when colloidal nano-objects are concerned, the reversibility or irreversibility are strongly dependent on the interfacial tension between the fluids, and an adsorption-desorption equilibrium, similar to that found in common surfactants, could possibly appear. Despite the importance of the contact angle, its experimental determination is far from trivial. Nowadays, there are different available techniques enabling its determination., which are extensively discussed in recent reviews by Maestro et al. [31] and Zanini and Isa [63].
There are several approaches for tuning the wettability of colloidal particles, which are based on the modification of the surface nature of the particles through chemical or physical procedures [27]. The former frequently consider the irreversible attachment of ligands onto the particle surface through a selective chemical reaction enabling the formation of covalent bonds, e.g., thiols onto gold surfaces or silanes onto SiO2 surfaces [64,65,66]. The modification of particle wettability through physical procedures relies on the interaction (through electrostatic, van der Waals, or hydrogen bonding interactions) of particles with surface-active molecules, generally surfactants or polymers [44,45,46], but in some cases low molecular weight compounds, such as alcohols, allow modifying the wettability of the particles [47,67].
The above discussion demonstrates that both the wettability of colloidal objects and the decrease of the interfacial tension play a central role in controlling the assembly of particles at the fluid interfaces. However, a complete description of the assembly process also requires including the role of the interactions between particles at the interface [24,27,33,68]. This is because the interactions involved in the assembly process, which can present different origins, for example, electrostatic, van der Waals, hydrogen bonds, and covalent bonds, controls the structure and dynamics within the interface, impacting, as matter of fact, in the physicochemical properties of the assembled systems, and consequently in their potential applications.
2.3. Organization of Colloids at Interfaces
The wettability of the colloidal particles and the interactions appearing within the interface [30,69,70] govern the structure and equilibrium properties of particle-laden interfaces. Several studies have shown that the hydrophobicity of the particles presents a strong impact on both the interfacial coverage and the position of the particles within the interfacial plane [44,71,72]. For example, Santini et al. [45] studied the adsorption of monolayers of silica nanoparticles decorated with palmitic acid at the water-vapor interface. Their results showed an enhanced interfacial coverage with particles hydrophobicity. On one hand, particles with the lowest hydrophobicity have been found to form isolated particle rafts at the interface, and on the other hand, the increase of the hydrophobicity of the particles has yielded the formation of close-packed films. The increase of hydrophobicity was clearly related to a sharp decrease of the interfacial tension and an increase of the layer thickness and the surface concentration of particles at the interface. Therefore, it is possible to assume that an increase of the hydrophobicity of the particles is the driving force for the transition between layers with a lost packing to close-packed particle-laden interfaces [42,43,73,74,75].
Finally, a direct visualization of single microparticles at the interface is possible. Currently, it is routine to analyze their individual positions using video microscopy. Following this approach, it has been possible to analyze the densification process of particle-laden interfaces, revealing the existence of transitions between different phases as the interfacial coverage is increased, similar to the scenario found in the corresponding three-dimensional (3D) counterparts. Furthermore, such phase transitions appear independently of the particles size, with the latter slightly shifting the threshold interfacial densities in which the phase transition occurs [69,70,76].
3. Interactions between Colloids Trapped at Fluid Interfaces
From a fundamental point of view, the interactions occurring between colloidal particles trapped at a fluid interface are different from those expected on the corresponding 3D counterpart systems. This is associated with the role of the interface as a confining environment. Thus, a colloidal particle trapped at a fluid interface is considered as an object attached to a fluctuating surface that separates two different phases, with the physicochemical properties (e.g., density, dielectric permittivity, and ionic strength) of such phases being, in general, significantly different. Furthermore, the properties of such discontinuity can be modified by factors such as the size and shape of the assembled objects, their charge, their wettability, and their surface chemistry [27,33,45,68,77,78,79]. These factors can have a combined effect on the behavior of the interface, making it difficult to introduce a quantitative description of the interactions involved in systems formed by colloids trapped at fluid interfaces. Here, a brief description of the main forces driving the assembly of colloidal particles is presented. From a broad view, it is possible to classify the forces into the following two groups: (i) direct interactions, which are intrinsically related to the nature of colloidal objects (size, shape, surface chemistry, charge, and roughness) and (ii) external interactions, associated with the presence of external fields, which act on individual objects or sets of them.
3.1. Direct Interactions
A description of the interactions occurring in colloids assembled at fluid interfaces is difficult due to the complex balance that exists between different types of repulsive and attractive interactions, which involved many objects (an example of the different types of interactions is schematized in Figure 2).
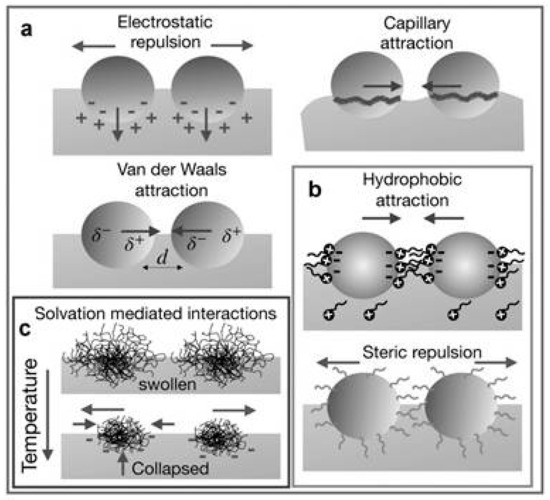
Figure 2.
Different types of interactions occurring between colloidal particles at a fluid interface: (a) Total electrostatic repulsions by combining dipolar and Coulomb interaction counterbalanced by van der Waals and capillary attractive forces, (b) ligand-mediated interactions, attractive hydrophobic forces and steric repulsions, and (c) solvation-mediated interactions that appear in particular cases due to changes in the environment. Adapted from Maestro [33], Copyright (2019), with permission from Elsevier.
The repulsive contribution is the result of a combination between a screened repulsive Coulombic interaction, which operates at short distances, and the long-range dipole–dipole interactions resulting from the asymmetric distribution of charges. This is associated with the different dissociation degree of the dissociable groups present at the colloid surface as a result of their immersion in fluids with different dipolar constants [80,81,82,83]. The role of the repulsive interaction is reinforced by steric contributions due to the presence of a shell surrounding the particle, which presents, in most of the cases, a polymeric nature. Thus, the overlapping of the shells of different particles leads to the emergence of an osmotic pressure that prevents the particles aggregation, avoiding the unfavorable decrease of the system entropy associated with the shell compression [84,85,86]. Considering the attractive interactions, it is necessary to mention the role of the van der Waals interaction. The impact of these interactions for colloids trapped at fluid interfaces is strongly influenced by the size, shape, and wettability of the objects [42,77]. Furthermore, the presence of hydrophobic interactions should not be neglected. These are the consequence of the requirement of minimization free energy as a result of the unfavorable nature of the contact between colloidal objects, with hydrophilic nature, and the solvent [87].
The last type of direct interactions which should be considered, when colloidal matter trapped at fluid interfaces is concerned, are the capillary forces. These can present both attractive and repulsive characteristics which appear from the interfacial deformation associated with the entrapment of the particles at the interface. This leads to the appearance of a meniscus which leads to the emergence of long-range interactions between colloids, even larger than the size of a single colloidal particle, to ensure the area minimization. The appearance of a meniscus around an isotropic colloid at a flat interface makes it necessary that the colloids exert a force in the direction perpendicular to the interface. This can occur when the densities of the colloidal particles and the fluid are similar, or when the colloids and the interface present charges [69,70,76,88,89,90,91]. In contrast, the role of capillary interactions can be considered negligible for small colloids, or when electrostatic interactions are weak. The appearance of a nonuniform wetting of the colloids for the interface can also lead to the appearance of a force in the direction perpendicular to the interface, distorting the contact line and enhancing the importance of the capillary interactions [92]. It is worth mentioning the important role of the interfacial heterogeneity in the interactions occurring within the interface. Particular cases of this are found when glass transitions appear in mixtures of particles with different sizes, or in the emergence of attractive interactions between colloids with charges that are equal are concerned. This can be explained by considering the asymmetric distribution of charge within the interface [37,76,93,94,95,96].
3.2. External Interactions
External fields have been revealed as a very effective way to tune the assembly of colloidal objects at interfaces, enabling control over the mechanical, optical, and electronic properties of these assembled systems [97,98]. The confinement of particles at fluid interfaces is associated with a restriction of their vertical motion, which is governed mainly by the wettability of the particles. It is expected that once the colloids are placed in their equilibrium positions, only small fluctuations appear around their equilibrium positions in relation to the interfacial place. However, when the motion within the interfacial plane is considered, particles can diffuse freely within such a plane [99]. This 2D diffusion can be manipulated by the mechanical deformation of the interface, by changing the interfacial area (dilatational deformations) or its shape (shear deformations), tuning the characteristic of the interfacial assembly which can even induce 2D phase transitions [100,101,102]. Similar effects can also be induced using external fields, e.g., magnetic or electric one [88,103].
Moreover, the change of the environmental conditions (pH, temperature, and ionic strength) can also be used to modify the interactions and the organization of particles at the fluid interface. Furthermore, this can also be exploited to tune the assembly of the particles at the interface, which has been explored mainly in the assembly of microgel particles [57,85,104].
The understanding of the interactions involving multiple bodies has special relevance because they can have a significant influence on the transport phenomena occurring within the confinement plane. Therefore, the modification of the energy landscape and the physicochemical properties of the interfaces have a critical impact on the development of new interface-based materials.
4. Dynamics of Colloids Trapped at Fluid Interfaces
The analysis of the dynamics of adsorbed colloid at the fluid interface needs to take into consideration the interactions involving several particles, as well as the interactions between the particles and the two adjacent fluids. Therefore, the motion of colloids trapped at the interface is strongly influenced by the existence of fluid phases, which controls, in many cases, the time scales [30,105].
The motion, in short time limits, for particles trapped at fluid-like monolayers which present compressible behavior, results in density fluctuations similar to that expected for a sound wave. This presents a characteristic time given by ts = R⁄cs, where R and cs are the radius of the particle and the speed of sound, respectively. These dynamics processes occur at extremely short times (~0.1 ns). Furthermore, a second dynamic defined by a characteristic time, th, appears associated with the hydrodynamic interactions. This latter process is coupled with the velocity field generated by the colloidal motion, which induces a drag force between the particles, appearing in a time scale close to 10 ns. This time scale is similar to the time needed for the displacement of the transverse moment generated by the particle motion, which is a distance similar to the interparticle distance. The hydrodynamics interactions can appear coupled to some additional processes associated with the initial velocity of the colloids, resulting from a time-dependent velocity field due to the particle motion. This type of motion presents a characteristic time associated with the transverse diffusion coefficient [30,106,107].
It is worth mentioning that the motion of the colloids depends on the diffusion coefficient , with being the Stokes friction coefficient. The characteristic time for colloidal motion at the interface can span from 1 ms to 1 s, which clearly shows evidence of the separation between the colloidal dynamics and other interfacial processes. Thus, it is possible to assume that the motion of a colloid can be described as a diffusion process affected for the interactions with neighboring colloids. Information about the dynamics of trapped particles can be obtained by analyzing the trajectories of colloids at the interface, which gives information on the mean square displacement (MSD, ), which is related to the diffusion coefficient, D, and the characteristic length of the translational motion [30,89,90,107,108],
where α is a scale exponent and d represent the number of dimensions. Considering a low interfacial coverage, linear dependence of the MSD would be expected on time, with the diffusion coefficient obtained from the slope of the representation defined by Equation (9) [30,107]. The increase of the coverage leads the system to a more complex dynamic, with the following two clear thresholds: (i) short times and (ii) long times. The existence of two regimes is characterized by a change of the time dependence of the MSD with the increase of the time, with such change being associated with the restriction of the diffusion region. At short times, the dynamics is limited to short distances (neighboring colloids), which leads to the appearance of the self-diffusion coefficient at short times,
The diffusion coefficient in the short time limit decreases with an increase of the interfacial coverage, , as a result of the interactions. Thus, , with being a parameter considering the interactions [109]. In the long time limit, the collective motions of the particles start being important for the interfacial dynamics, leading to a diffusion coefficient defined as
Considering the long time limit, the motion of a particle can be described in terms of the escaping of the particle from a box, which is defined by its close neighbors, i.e., the -relaxation [110]. At high interfacial coverages, the interparticle repulsion arrests the dynamics within the experimental window [90]. Therefore, the motion of the particles must be considered in terms of an harmonic Brownian oscillator (BHO), with the Langevin equation including a restoring elastic force accounting for the behavior of the particles [111]
where m and v correspond to the mass and velocity of one particle, respectively, and k is the force constant, with F(t) being a random force. Assuming the overdamped limit and neglecting the role of the inertia, the following solution is obtained [112]:
where . The characteristic time is defined as . A better description of the dynamics of Brownian particles when interactions are involved makes it necessary to introduce ad hoc corrections to Equation (13). This can be done considering the non-exponential character of the relaxation [113].
where c is a stretching exponent, assuming values below 1 and providing information about the width of the relaxation time spectrum. In Equation (14), a term giving information about the long time dynamics of particles is included. This term assumes the escaping dynamics of particles from their groups, resulting in the appearance of a linear dependence with time. For solid-like monolayers, it is necessary to introduce the overdamped bead-spring model (OBS) accounting for the dynamics when the coverage is high [114]
where accounts for the displacement of a particle i in a time t, with n being the number of closest neighbors to i. The high interfacial coverage existing in solid-like monolayers makes impossible the appearance of escaping dynamics, leading to the disappearance of the linear dependence of the MSD when long times are concerned. Thus, the solution reads as follows:
Equation (15) only considers the interaction with the closest neighbors, where represents the net factor and is the b-order wave-vector, whereas i represents a vector going from the particle i to the particle j. This model is reduced to the BHO one in the initial stages and tends to the following limit at long times:
where the threshold value depends on the strength of the harmonic force and the net factor.
5. Rheological Behavior of Particles Attached at Fluid Interfaces
Rheology studies the deformation and flow of materials as a response to the application of a mechanical disturbance. This can lead to a change in the material size (dilation) or in its shape (shear). The understanding of the relaxation processes as a result of mechanical deformation is of central importance for describing the mechanical behavior of materials, and their potential use in the fabrication of systems with technological interest [51,115,116,117,118]. This has stimulated extensive research activity aimed to shed light on the behavior of colloidal particles confined in 2D, which is of particular interest to the understanding of biological processes involving self-assembled systems operating under continuous mechanical perturbations.
Therefore, the understanding of the interfacial response against mechanical perturbations transcends the field of physicochemical, and should be considered a challenge with a marked multidisciplinary nature [119,120,121,122,123,124,125,126,127,128,129,130]. Figure 3 schematized the different deformation modes that can undergo a fluid interface, including those occurring within the plane (dilation and shear) and those occurring out the plane (bending and splaying). In the following, some of the main aspects associated with the response of particle-laden interface against in-plane mechanical perturbations, either dilational or shear one, are analyzed.
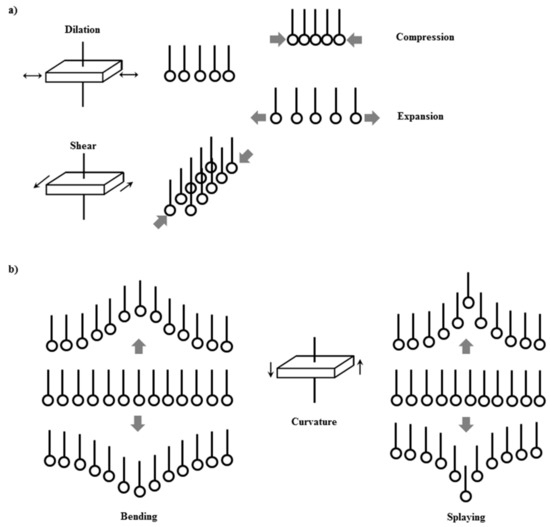
Figure 3.
Sketch of the different dynamic modes appearing for fluid interfaces: (a) In-plane modes (dilation and shear) and (b) out-of-plane modes (bending and splaying).
5.1. Dilational Rheology
The dilational rheology of interfaces gives information related to changes in surface tension as result of changes in the interfacial are, or what is the same in the interfacial coverage. Therefore, a time-dependent infinitesimal change of the interfacial, δA(t), due to an uniaxial deformation, produces a change in surface pressure, δΠ(t), defined as [131,132]:
where represents the time-dependent viscoelastic modulus. Equation (17) gives a generic definition considering a surface pressure change associated with the modification of the surface area . Under equilibrium conditions, the dynamics modulus assumes a value similar to that corresponding to the Gibbs elasticity
where represents the surface excess. Assuming a small amplitude deformation with a characteristic frequency ω, the complex modulus is defined as,
where is the storage modulus, the imaginary part the loss modulus is , and represents the dilational interfacial viscosity. From the above presented definition of the dilatational viscoelastic modulus, it is expected that such a parameter can be changed either from the adsorption of colloids at the interface or from the modification of the available interfacial areas, which results in interfacial relaxation processes occurring in different time scales [46]. Despite the many efforts made to understand the behavior of particle monolayers against dilational deformations, there are several aspects that still remain unclear, especially those related to the theoretical description of the experimental results [52,53,133,134]. The first theoretical description of the mechanical response of interfaces with particles was given by the Miller group [52,53], who followed a similar procedure to that used in the description of proteins, and mixtures of proteins and surfactants [54,135], at fluid interfaces, gave a description of the mechanical response of particle-laden interfaces. However, such description is merely theoretical, without any development of its practical application.
It has been mentioned that the adsorption of particles at fluid interfaces is strongly influenced by its wettability, and thus some effect of this property can be expected in the mechanical performance of the interface, as was demonstrated by Safouane et al. [136]. Thus, the increase of the hydrophobicity of the particles favors their incorporation at the interface, resulting in layers with higher values of the storage modulus than that corresponding to the loss one. This behavior is in turn related to an increase in the interactions between particles at the interface [137]. Analyzing the characteristic relaxation process, it was found that particles undergo a reorganization process within the interface (~10−3 s). Such relaxation was slowed down by the increase of the hydrophobicity of the particles due to the increase of the steric hindrance at the interface [138].
Polymeric particles present a behavior slightly different to that discussed above as a result of their partially deformable character, with both the storage and the loss moduli increasing with the interfacial coverage. However, above a threshold value of the interfacial coverage, a steep decrease of the viscoelastic moduli occurs due to the possible distortion of the quasi-2D layer, appearing as the buckling of the monolayer. Such systems present a transition between a fluid-like behavior to a solid-like behaviour with the densification of the interface [69,139,140]. Therefore, we assume that the dilational rheological response is strongly dependent on the specific region of the phase diagram under study [73]. For polymeric particles, the storage modulus was higher than the loss modulus within the entire interfacial coverage range, with this behavior being independent on the stain rate [139]. For low interfacial densities, the elastic component increases with the interfacial coverage due to the presence of repulsive interactions. When such repulsions are screened, an increase of both storage and loss moduli was found with the interfacial coverage until the formation of a close-packed monolayer [73]. For the highest interfacial densities, values of the storage modulus in the range of 350–600 mN/m were found.
5.2. Shear Rheology
For deformations in the interfacial plane, the shear elasticity can be defined as a constant of proportionality between the applied stress, uxy, and the resulting deformation, σxy. For a solid monolayer, such proportionality can be expressed in terms of Hook’s Law, σxy = Guxy. However, when fluid-like monolayers are concerned, it is the viscosity that defines the behavior, σxy = η(duxy/dt), where η represents the interfacial viscosity and (duxy/dt) the strain rate. For oscillatory deformations, the complex viscoelastic modulus can be defined as,
where and are the storage and loss moduli, respectively. The first study focused on the response against shear deformations of particle-laden interfaces was performed by Cicuta et al. [141]. They found that the reduced deformability of the particles leads to a behavior controlled by the loss modulus (viscous behavior) which contrasts with the elastic response found when deformable molecules are concerned. Furthermore, a sharp increase of the viscoelastic modulus was found by the interfacial coverage. Thus, the behavior of quasi-2D systems against shear deformations appears significantly to that found for their respective 3D counter-parts, as was also independently stated by Trappe et al. [142]. Further developments of the studies related to the shear response of particle-laden interfaces evidence the critical role of the interfacial organization of the particles in the interfacial shear response, with behavior for systems in which aggregation appears similar that that found in 3D systems. This is explained by the influence of the interactions in the rheological response of interfaces [143].
For asymmetric particles, it was found that the anisotropic reorganization of particles at the interface plays a central role on the shear response, with a behavior reminiscent of a 2D-plasticized system when the coverage is low. The increase of the coverage leads to a perfect elastic interfacial behavior which differs from the scenario found for spherical particles [144]. Barman and Cristopher [145] found that the increase of the interfacial coverage drives a transition between shear-thinning to yielding one as a result of the differences in the stresses dissipation appearing for asymmetric particles. Furthermore, the linearity of the response disappears with the increase of the interfacial density, with the viscoelastic modulus presenting a power-law dependence on the surface coverage [142]. Madivala et al. [146] found that the strength of the electrostatic interactions between asymmetric particles defines the shear response of the particle-laden interface, with the interfacial organization controlling the interfacial properties. The role of the morphology of the particles was confirmed using non-spherical particles with different aspect ratio, which showed shear-thickening response. Furthermore, the increase of the particles asymmetry decreases the interfacial density in which jamming occurs [147].
The particle roughness also plays an important role in the rheological response of particle-laden interface, presenting effect that depends on the interfacial coverage. For low interfacial coverage, the roughness reduces the shear viscosity, with the opposite being true when the interfacial density increases. This is the result of the decrease of the interparticle friction which influences the role of particles in emulsion stabilization [147,148].
It is expected that the contact angle of particles trapped at the fluid interface also influences the rheological response. Safouane et al. [136] found that an increase in particle hydrophobicity leads to the increase of storage and loss moduli, with the monolayers of particles presenting the lowest hydrophobicity having a mainly elastic behavior. The increase of the hydrophobicity leads to an increase of the viscous character, with particles presenting a contact angle around 90° having a gel-like behavior (G′ = G″)
6. Conclusions
This review reports some of the most relevant physicochemical bases underlying the attachment of colloidal systems to fluid interfaces. The prospects of tuning and controlling the assembly of colloids at interfaces following different approaches, such as modifying particle’s wettability, intermolecular interactions and interfacial flows, make them potential candidates to create complex and hierarchical structures that have many potential technological applications. There are to date many experimental and theoretical efforts to unravel the complex physics involved in the interfacial assembly of colloidal particles. However, to obtain a complete framework describing the behavior of particle-laden interfaces is still a challenge. This is due to the existence of many parameters (shape and size of the particles, chemical nature, wettability, etc.) affecting the interfacial morphology and response against mechanical deformations, which can influence in the future applications of particle-laden interfaces.
Author Contributions
The contributions of A.M. and E.G. have been equal in the different aspects of the work reported.
Funding
This research was funded by MINECO; grant number CTQ2016-78895-R and by Banco Santander-Universidad Complutense grant PR87/19-22513.
Acknowledgments
Authors are indebted to R.G. Rubio and F. Ortega for their friendship, continuous support, and encouragement in the development of their research careers. The C.A.I. of Espectroscopia y Correlación of the Universidad Complutense de Madrid is acknowledged for its availability in the use of its facilities.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Shi, S.; Russel, T.P. Nanoparticle Assembly at Liquid–Liquid Interfaces: From the Nanoscale to Mesoscale. Adv. Mater. 2018, 30, 1800714. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.; Kim, P.Y.; Xie, G.; Liu, X.; Helms, B.A.; Russel, T.P. Building Reconfigurable Devices Using Complex Liquid-Fluid Interfaces. Adv. Mater. 2019, 31, 1806370. [Google Scholar] [CrossRef] [PubMed]
- Garbin, V. Colloidal particles: Surfactants with a difference. Phys. Today 2013, 66, 67–68. [Google Scholar] [CrossRef]
- Binks, B.P. Particles as surfactants—Similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- Luengo, G.S.; Galliano, A.; Dubief, C. Aqueous Lubrication in Cosmetic. In Aqueous Lubrication. Natural and Biomimetic Approaches; Spencer, N.D., Ed.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2014; pp. 103–144. [Google Scholar]
- Llamas, S.; Guzmán, E.; Ortega, F.; Baghdadli, N.; Cazeneuve, C.; Rubio, R.G.; Luengo, G.S. Adsorption of polyelectrolytes and polyelectrolytes-surfactant mixtures at surfaces: A physico-chemical approach to a cosmetic challenge. Adv. Colloid Interface Sci. 2015, 222, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F.; Wennerström, H. The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet; Wiley-VCH: Hoboken, NJ, USA, 1994. [Google Scholar]
- Morrison, I.D.; Ross, S. Colloidal Dispersions: Suspensions, Emulsions, and Foams; Wiley-Interscience: Hoboken, NJ, USA, 2002. [Google Scholar]
- Santini, E.; Guzmán, E.; Ferrari, M.; Liggieri, L. Emulsions stabilized by the interaction of silica nanoparticles and palmitic acid at the water–hexane interface. Colloids Surf. A 2014, 460, 333–341. [Google Scholar] [CrossRef]
- Maestro, A.; Rio, E.; Drenckhan, W.; Langevin, D.; Salonen, A. Foams stabilised by mixtures of nanoparticles and oppositely charged surfactants: Relationship between bubble shrinkage and foam coarsening. Soft Matter 2014, 10, 6975–6983. [Google Scholar] [CrossRef]
- Arriaga, L.R.; Drenckhan, W.; Salonen, A.; Rodrigues, J.A.; Íñiguez-Palomares, R.; Rio, E.; Langevin, D. On the long-term stability of foams stabilised by mixtures of nano-particles and oppositely charged short chain surfactants. Soft Matter 2012, 8, 11085–11097. [Google Scholar] [CrossRef]
- Santini, E.; Ravera, F.; Ferrari, M.; Alfè, M.; Ciajolo, A.; Liggieri, L. Interfacial properties of carbon particulate-laden liquid interfaces and stability of related foams and emulsions. Colloids Surf. A 2010, 365, 189–198. [Google Scholar] [CrossRef]
- Santini, E.; Guzmán, E.; Ravera, F.; Ciajolo, A.; Alfè, M.; Liggieri, L.; Ferrari, M. Soot particles at the aqueous interface and effects on foams stability. Colloids Surf. A 2012, 413, 216–223. [Google Scholar] [CrossRef]
- Carter, B.O.; Wang, W.; Bray, C.L.; Adams, D.J.; Cooper, A.I. Methane Storage in Dry Water Gas Hydrates. J. Am. Chem. Soc. 2008, 130, 11608–11609. [Google Scholar]
- Nguyen, A.; Schulze, H.J. Colloidal Science of Flotation; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Asuri, P.; Karajanagi, S.S.; Dordick, J.S.; Kane, R.S. Directed Assembly of Carbon Nanotubes at Liquid−Liquid Interfaces: Nanoscale Conveyors for Interfacial Biocatalysis. J. Am. Chem. Soc. 2006, 128, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Varadaraj, R. Colloid and interface science in the oil industry. Curr. Opin. Colloid Interface Sci. 1999, 1, 535–539. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef]
- Guzmán, E.; Santini, E. Lung surfactant-particles at fluid interfaces for toxicity assessments. Curr. Opin. Colloid Interface Sci. 2019, 39, 24–39. [Google Scholar] [CrossRef]
- Hinman, S.S.; McKeating, K.S.; Cheng, Q. Surface Plasmon Resonance: Material and Interface Design for Universal Accessibility. Anal. Chem. 2018, 90, 19–39. [Google Scholar] [CrossRef]
- Brilson, L.J. An Essential Guide to Electronic Material Surfaces and Interfaces; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Hari Babu Vasili, H.B.; Gamino, M.; Gàzquez, J.; Sánchez, F.; Valvidares, M.; Gargiani, P.; Pellegrin, E.; Fontcuberta, J. Magnetoresistance in Hybrid Pt/CoFe2O4 Bilayers Controlled by Competing Spin Accumulation and Interfacial Chemical Reconstruction. ACS Appl. Mater. Interfaces 2018, 10, 12031–12041. [Google Scholar] [CrossRef]
- Ballauf, M. Self-assembly creates 2D materials. Science 2016, 352, 656–657. [Google Scholar] [CrossRef]
- Maestro, A.; Santini, E.; Guzmán, E. Physico-chemical foundations of particle-laden fluid interfaces. Eur. Phys. J. E 2018, 41, 97. [Google Scholar] [CrossRef]
- Maestro, A.; Santini, E.; Zabiegaj, D.; Llamas, S.; Ravera, F.; Liggieri, L.; Ortega, F.; Rubio, R.G.; Guzman, E. Particle and Particle-Surfactant Mixtures at Fluid Interfaces: Assembly, Morphology, and Rheological Description. Adv. Condens. Matter Phys. 2015, 2015, 917516. [Google Scholar] [CrossRef]
- Garbin, V. Collapse mechanisms and extreme deformation of particle-laden interfaces. Curr. Opin. Colloid Interface Sci. 2019, 39, 202–211. [Google Scholar] [CrossRef]
- Garbin, V.; Crocker, J.C.; Stebe, K.J. Nanoparticles at fluid interfaces: Exploiting capping ligands to control adsorption, stability and dynamics. J. Colloid Interface Sci. 2012, 387, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.; Llamas, S.; Maestro, A.; Fernández-Peña, L.; Akanno, A.; Miller, R.; Ortega, F.; Rubio, R.G. Polymer–surfactant systems in bulk and at fluid interfaces. Adv. Colloid Interface Sci. 2016, 233, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Guzmán, E.; Santini, E. Interfacial Rheology of Particle-Laden Interfaces and Its Role in the Stabilization of Dispersed Systems. In Rheology: Principles, Applications and Environmental Impacts; Karpushkin, E., Ed.; Nova Science Publisher, Inc.: New York, NJ, USA, 2015; pp. 1–26. [Google Scholar]
- Mendoza, A.J.; Guzmán, E.; Martínez-Pedrero, F.; Ritacco, H.; Rubio, R.G.; Ortega, F.; Starov, V.M.; Miller, R. Particle laden fluid interfaces: Dynamics and interfacial rheology. Adv. Colloid Interface Sci. 2014, 206, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Guzmán, E.; Ortega, F.; Rubio, R.G. Contact angle of micro- and nanoparticles at fluid interfaces. Curr. Opin. Colloid Interface Sci. 2014, 19, 355–367. [Google Scholar] [CrossRef]
- Guzmán, E.; Santini, E.; Liggieri, L.; Ravera, F.; Loglio, G.; Maestro, A.; Rubio, R.G.; Krägel, J.; Grigoriev, D.; Miller, R. Particle-Surfactant Interaction at Liquid Interfaces. In Colloid and Interface Chemistry for Nanotechnology; Kralchevsky, P., Miller, R., Ravera, F., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 77–109. [Google Scholar]
- Maestro, A. Tailoring the interfacial assembly of colloidal particles by engineering the mechanical properties of the interface. Curr. Opin. Colloid Interface Sci. 2019, 39, 232–250. [Google Scholar] [CrossRef]
- Thijssen, J.H.J.; Vermant, J. Interfacial rheology of model particles at liquid interfaces and its relation to (bicontinuous) Pickering emulsions. J. Phys. Condens. Matter 2017, 30, 02300. [Google Scholar] [CrossRef]
- Dasgupta, S.; Auth, T.; Gompper, G. Nano- and microparticles at fluid and biological interfaces. J. Phys. Condens. Matter 2017, 29, 373003. [Google Scholar] [CrossRef]
- Varga, I.; Campbell, R.A. General Physical Description of the Behavior of Oppositely Charged Polyelectrolyte/Surfactant Mixtures at the Air/Water Interface. Langmuir 2017, 33, 5915–5924. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, M.A.; Chen, L.; Deming, C.P.; Rodriguez-Valverde, M.A.; Chen, S.; Cabrerizo-Vilchez, M.A.; Hidalgo-Alvarez, R. A simple strategy to improve the interfacial activity of true Janus gold nanoparticles: A shorter hydrophilic capping ligand. Soft Matter 2016, 12, 31–34. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, M.A.; Ramos, J.; Isa, L.; Rodriguez-Valverde, M.A.; Cabrerizo-Vilchez, M.A.; Hidalgo-Alvarez, R. Interfacial Activity and Contact Angle of Homogeneous, Functionalized, and Janus Nanoparticles at the Water/Decane Interface. Langmuir 2015, 31, 8818–8823. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rodriguez, M.A.; Song, Y.; Rodríguez-Valverde, M.Á.; Chen, S.; Cabrerizo-Vilchez, M.A.; Hidalgo-Alvarez, R. Comparison of the Interfacial Activity between Homogeneous and Janus Gold Nanoparticles by Pendant Drop Tensiometry. Langmuir 2014, 30, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Noskov, B.A.; Bykov, A.G. Dilational rheology of monolayers of nano- and micropaticles at the liquid-fluid interfaces. Curr. Opin. Colloid Interface Sci. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Yazhgur, P.A.; Noskov, B.A.; Liggieri, L.; Lin, S.Y.; Loglio, G.; Miller, R.; Ravera, F. Dynamic properties of mixed nanoparticle/surfactant adsorption layers. Soft Matter 2013, 9, 3305–3314. [Google Scholar] [CrossRef]
- Maestro, A.; Guzmán, E.; Santini, E.; Ravera, F.; Liggieri, L.; Ortega, F.; Rubio, R.G. Wettability of silica nanoparticle–surfactant nanocomposite interfacial layers. Soft Matter 2012, 8, 837–843. [Google Scholar] [CrossRef]
- Santini, E.; Guzmán, E.; Ravera, F.; Ferrari, M.; Liggieri, L. Properties and structure of interfacial layers formed by hydrophilic silica dispersions and palmitic acid. Phys. Chem. Chem. Phys. 2012, 14, 607–615. [Google Scholar] [CrossRef]
- Liggieri, L.; Santini, E.; Guzmán, E.; Maestro, A.; Ravera, F. Wide-frequency dilational rheology investigation of mixed silica nanoparticle–CTAB interfacial layers. Soft Matter 2011, 7, 7699–7709. [Google Scholar] [CrossRef]
- Zabiegaj, D.; Santini, E.; Guzmán, E.; Ferrari, M.; Liggieri, L.; Ravera, F. Carbon Soot-Ionic Surfactant Mixed Layers at Water/Air Interfaces. J. Nanosci. Nanotechnol. 2015, 15, 3618–3625. [Google Scholar] [CrossRef]
- Zabiegaj, D.; Santini, E.; Guzmán, E.; Ferrari, M.; Liggieri, L.; Buscaglia, V.; Buscaglia, M.T.; Battilana, G.; Ravera, F. Nanoparticle laden interfacial layers and application to foams and solid foams. Colloids Surf. A 2013, 438, 132–140. [Google Scholar] [CrossRef]
- Llamas, S.; Mendoza, A.J.; Guzmán, E.; Ortega, F.; Rubio, R.G. Salt effects on the air/solution interfacial properties of PEO-containing copolymers: Equilibrium, adsorption kinetics and surface rheological behavior. J. Colloid Interface Sci. 2013, 400, 49–58. [Google Scholar] [CrossRef]
- Llamas, S.; Fernández-Peña, L.; Akanno, A.; Guzmán, E.; Ortega, V.; Ortega, F.; Csaky, A.G.; Campbell, R.A.; Rubio, R.G. Towards understanding the behavior of polyelectrolyte–surfactant mixtures at the water/vapor interface closer to technologically-relevant conditions. Phys. Chem. Chem. Phys. 2018, 20, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Akanno, A.; Guzmán, E.; Fernández-Peña, L.; Llamas, S.; Ortega, F.; Rubio, R.G. Equilibration of a Polycation–Anionic Surfactant Mixture at the Water/Vapor Interface. Langmuir 2018, 34, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Aksenenko, E.V.; Fainerman, V.B. Dynamic interfacial tension of surfactant solutions. Adv. Colloid Interface Sci. 2017, 247, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Akanno, A.; Guzmán, E.; Ortega, L.F.-P.F.; Rubio, R.G. Surfactant-Like Behavior for the Adsorption of Mixtures of a Polycation and Two Different Zwitterionic Surfactants at theWater/Vapor Interface. Molecules 2019, 24, 3442. [Google Scholar] [CrossRef] [PubMed]
- Fainerman, V.B.; Kovalchuk, V.I.; Lucassen-Reynders, E.H.; Grigoriev, D.O.; Ferri, J.K.; Leser, M.E.; Michel, M.; Miller, R.; Mohwald, H. Surface-Pressure Isotherms of Monolayers Formed by Microsize and Nanosize Particles. Langmuir 2006, 22, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Fainerman, V.B.; Kovalchuk, V.I.; Grigoriev, D.O.; Leser, M.E.; Michel, M. Composite interfacial layers containing micro-size and nano-size particles. Adv. Colloid Interface Sci. 2006, 128–130, 17–26. [Google Scholar] [CrossRef]
- Miller, R.; Fainerman, V.B.; Makievski, A.V.; Kragel, J.; Grigoriev, D.O.; Kazakov, V.N.; Sinyachenko, O.V. Dynamics of protein and mixed proteinr/surfactant adsorption layers at the water/fluid interface. Adv. Colloid Interface Sci. 2000, 86, 39–82. [Google Scholar] [CrossRef]
- Groot, R.; Stoyanov, S. Equation of state of surface-adsorbing colloids. Soft Matter 2011, 6, 1682–1692. [Google Scholar] [CrossRef]
- Mulero, A. (Ed.) Theory and Simulation of Hard-Sphere Fluids and Related System; Springer: Berlin, Germany, 2008. [Google Scholar]
- Deshmukh, O.S.; Maestro, A.; Duits, M.H.G.; van den Ende, D.; Stuart, M.C.; Mugele, F. Equation of state and adsorption dynamics of soft microgel particles at an air–water interface. Soft Matter 2014, 10, 7045–7050. [Google Scholar] [CrossRef]
- Levine, S.; Bowen, B.D.; Partridge, S.J. Stabilization of emulsions by fine particles I. Partitioning of particles between continuous phase and oil/water interface. Colloids Surf. 1989, 38, 325–344. [Google Scholar] [CrossRef]
- Zang, D.Y.; Rio, E.; Delon, G.; Langevin, D.; Wei, B.; Binks, B.P. Influence of the contact angle of silica nanoparticles at the air–water interface on the mechanical properties of the layers composed of these particles. Mol. Phys. 2011, 109, 1057–1066. [Google Scholar] [CrossRef]
- Stocco, A.; Rio, E.; Binks, B.P.; Langevin, D. Aqueous foams stabilized solely by particles. Soft Matter 2011, 7, 1260–1267. [Google Scholar] [CrossRef]
- Herzig, E.M.; White, K.A.; Schofield, A.B.; Poon, W.C.K.; Clegg, P.S. Bicontinuous emulsions stabilized solely by colloidal particles. Nat. Mater. 2007, 6, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Danov, K.D.; Krachevsky, P.A. Capillary forces between particles at a liquid interface: General theoretical approach and interactions between capillary multipoles. Adv. Colloid Interface Sci. 2010, 154, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Zanini, M.; Isa, L. Particle contact angles at fluid interfaces: Pushing the boundary beyond hard uniform spherical colloids. J. Phys. Condens. Matter 2016, 28, 313002. [Google Scholar] [CrossRef]
- Fletcher, P.D.I.; Holt, B.L. Controlled Silanization of Silica Nanoparticles to Stabilize Foams, Climbing Films, and Liquid Marbles. Langmuir 2011, 27, 12869–12876. [Google Scholar] [CrossRef]
- Petcu, C.; Purcar, V.; Spătaru, C.-I.; Alexandrescu, E.; Şomoghi, R.; Trică, B.; Niţu, S.G.; Panaitescu, D.M.; Donescu, D.; Jecu, M.-L. The Influence of New Hydrophobic Silica Nanoparticles on the Surface Properties of the Films Obtained from Bilayer Hybrids. Nanomaterials 2017, 7, 47. [Google Scholar] [CrossRef]
- Guzmán, E.; Ortega, F.; Baghdadli, N.; Cazeneuve, C.; Luengo, G.S.; Rubio, R.G. Adsorption of Conditioning Polymers on Solid Substrates with Different Charge Density. ACS Appl. Mater. Interfaces 2011, 3, 3181–3188. [Google Scholar] [CrossRef]
- Maestro, A.; Bonales, L.J.; Ritacco, H.; Rubio, R.G.; Ortega, F. Effect of the spreading solvent on the three-phase contact angle of microparticles attached at fluid interfaces. Phys. Chem. Chem. Phys. 2010, 12, 14115–14120. [Google Scholar] [CrossRef]
- Bresme, F.; Oettel, M. Nanoparticles at fluid interfaces. J. Phys. Condens. Matter 2007, 19, 413101. [Google Scholar] [CrossRef]
- Bonales, L.J.; Rubio, J.E.F.; Ritacco, H.; Vega, C.; Rubio, R.G.; Ortega, F. Freezing Transition and Interaction Potential in Monolayers of Microparticles at Fluid Interfaces. Langmuir 2011, 27, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Parolini, L.; Law, A.D.; Maestro, A.; Martin, D.; Buzza, A.; Cicuta, P. Interaction between colloidal particles on an oil–water interface in dilute and dense phases. J. Phys. Condens. Matter 2015, 27, 194119. [Google Scholar] [CrossRef] [PubMed]
- Zang, D.; Stocco, A.; Langevin, D.; Wei, B.; Binks, B.P. An ellipsometry study of silica nanoparticle layers at the water surface. Phys. Chem. Chem. Phys. 2009, 11, 9522–9529. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Krägel, J.; Ravera, F.; Liggieri, L.; Miller, R. Study of the monolayer structure and wettability properties of silica nanoparticles and CTAB using the Langmuir trough technique. Colloids Surf. A 2011, 382, 186–191. [Google Scholar] [CrossRef]
- Bykov, A.G.; Noskov, B.A.; Loglio, G.; Lyadinskaya, V.V.; Miller, R. Dilational surface elasticity of spread monolayers of polystyrene microparticles. Soft Matter 2014, 10, 6499–6505. [Google Scholar] [CrossRef]
- Noskov, B.A.; Yazhgur, P.A.; Liggieri, L.; Lin, S.-Y.; Loglio, G.; Miller, R.; Ravera, F. Dilational rheology of spread and adsorbed layers of silica nanoparticles at the liquid-gas interface. Colloid J. 2014, 76, 127–138. [Google Scholar] [CrossRef]
- Bykov, A.G.; Loglio, G.; Miller, R.; Noskov, B.A. Dilational surface elasticity of monolayers of charged polystyrene nano- and microparticles at liquid/fluid interfaces. Colloids Surf. A 2015, 485, 42–48. [Google Scholar] [CrossRef]
- Bonales, L.J.; Martínez-Pedrero, F.; Rubio, M.A.; Rubio, R.G.; Ortega, F. Phase Behavior of Dense Colloidal Binary Monolayers. Langmuir 2012, 28, 16555–16566. [Google Scholar] [CrossRef]
- Girotto, M.; dos Santos, A.P.; Levin, Y. Interaction of Charged Colloidal Particles at the Air–Water Interface. J. Phys. Chem. B 2016, 120, 5817–5822. [Google Scholar] [CrossRef]
- Bresme, F.; Lehle, H.; Oettel, M. Solvent-mediated interactions between nanoparticles at fluid interfaces. J. Chem. Phys. 2009, 130, 214711. [Google Scholar] [CrossRef]
- Oettel, M.; Dietrich, S. Colloidal Interactions at Fluid Interfaces. Langmuir 2008, 24, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Pieranski, P. Two-Dimensional Interfacial Colloidal Crystals. Phys. Rev. Lett. 1980, 45, 569–572. [Google Scholar] [CrossRef]
- Hurd, A.J. The electrostatic interaction between interfacial colloidal particles. J. Phys. A Math. Gen. 1985, 18, L1055–L1060. [Google Scholar] [CrossRef]
- Masschaele, K.; Park, B.J.; Furst, E.M.; Fransaer, J.; Vermant, J. Finite Ion-Size Effects Dominate the Interaction between Charged Colloidal Particles at an Oil-Water Interface. Phys. Rev. Lett. 2010, 105, 048303. [Google Scholar] [CrossRef] [PubMed]
- Jun Park, B.; Vermant, J.; Furst, E.M. Heterogeneity of the electrostatic repulsion between colloids at the oil–water interface. Soft Matter 2010, 6, 5327–5333. [Google Scholar] [CrossRef]
- Birdi, K.S. (Ed.) Handbook of Surface and Colloid Chemistry; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Maestro, A.; Jones, D.; Sánchez de Rojas Candela, C.; Guzman, E.; Duits, M.H.G.; Cicuta, P. Tuning Interfacial Properties and Processes by Controlling the Rheology and Structure of Poly(N-isopropylacrylamide) Particles at Air/Water Interfaces. Langmuir 2018, 34, 7067–7076. [Google Scholar] [CrossRef]
- Maestro, A.; Guzmán, E.; Chuliá, R.; Ortega, F.; Rubio, R.; Miller, R. Fluid to soft-glass transition in a quasi-2D system: Thermodynamic and rheological evidences for a Langmuir monolayer. Phys. Chem. Chem. Phys. 2011, 13, 9534–9549. [Google Scholar] [CrossRef]
- Tabor, R.F.; Grieser, F.; Dagastine, R.R.; Chan, D.Y.C. The hydrophobic force: Measurements and methods. Phys. Chem. Chem. Phys. 2014, 16, 18065–18075. [Google Scholar] [CrossRef]
- Wickman, H.H.; Korley, J.N. Colloid crystal self-organization and dynamics at the air/water interface. Nature 1998, 393, 445–447. [Google Scholar] [CrossRef]
- Orsi, D.; Rimoldi, T.; Guzmán, E.; Liggieri, L.; Ravera, F.; Ruta, B.; Cristofolini, L. Hydrophobic Silica Nanoparticles Induce Gel Phases in Phospholipid Monolayers. Langmuir 2016, 32, 4868–4876. [Google Scholar] [CrossRef]
- Orsi, D.; Guzmán, E.; Liggieri, L.; Ravera, F.; Ruta, B.; Chushkin, Y.; Rimoldi, T.; Cristofolini, L. 2D dynamical arrest transition in a mixed nanoparticle-phospholipid layer studied in real and momentum spaces. Sci. Rep. 2015, 5, 17930. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.; Orsi, D.; Cristofolini, L.; Liggieri, L.; Ravera, F. Two-Dimensional DPPC Based Emulsion-like Structures Stabilized by Silica Nanoparticles. Langmuir 2014, 30, 11504–11512. [Google Scholar] [CrossRef]
- Loudet, J.-C.; Alsayed, A.M.; Zhang, J.; Yodh, A.G. Capillary Interactions Between Anisotropic Colloidal Particles. Phys. Rev. Lett. 2005, 94, 018301. [Google Scholar] [CrossRef] [PubMed]
- Uppapalli, S.; Zhao, H. The influence of particle size and residual charge on electrostatic interactions between charged colloidal particles at an oil–water interface. Soft Matter 2014, 10, 4555–4560. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rodriguez, M.A.; Binks, B.P.; Rodriguez-Valverde, M.A.; Cabrerizo-Vilchez, M.A.; Hidalgo-Alvarez, R. Particles adsorbed at various non-aqueous liquid-liquid interfaces. Adv. Colloid Interface Sci. 2017, 247, 208–222. [Google Scholar] [CrossRef]
- Montes Ruiz-Cabello, F.J.; Moazzami-Gudarzi, M.; Elzbieciak-Wodka, M.; Maroni, P.; Labbez, C.; Borkovec, M.; Trefalt, G. Long-ranged and soft interactions between charged colloidal particles induced by multivalent coions. Soft Matter 2015, 11, 1562–1571. [Google Scholar] [CrossRef]
- Botto, L.; Lewandowski, E.P.; Cavallaro, M.; Stebe, K.J. Capillary interactions between anisotropic particles. Soft Matter 2012, 8, 9957–9971. [Google Scholar] [CrossRef]
- Grzelczak, M.; Vermant, J.; Furst, E.M.; Liz-Marzán, L.M. Directed Self-Assembly of Nanoparticles. ACS Nano 2010, 4, 3591–3605. [Google Scholar] [CrossRef]
- Giner-Casares, J.J.; Reguera, J. Directed self-assembly of inorganic nanoparticles at air/liquid interfaces. Nanoscale 2016, 8, 16589–16595. [Google Scholar] [CrossRef]
- Dörr, A.; Hardt, S.; Masoud, H.; Stone, H.A. Drag and diffusion coefficients of a spherical particle attached to a fluid–fluid interface. J. Fluid Mech. 2016, 790, 607–618. [Google Scholar] [CrossRef]
- Erni, P. Deformation modes of complex fluid interfaces. Soft Matter 2011, 7, 7586–7600. [Google Scholar] [CrossRef]
- Maestro, A.; Zaccone, A. Nonaffine deformation and tunable yielding of colloidal assemblies at the air–water interface. Nanoscale 2017, 9, 18343–18351. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Deshmukh, O.S.; Mugele, F.; Langevin, D. Interfacial Assembly of Surfactant-Decorated Nanoparticles: On the Rheological Description of a Colloidal 2D Glass. Langmuir 2015, 31, 6289–6297. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pedrero, F.; Benet, J.; Rubio, J.E.F.; Sanz, E.; Rubio, R.G.; Ortega, F. Field-induced sublimation in perfect two-dimensional colloidal crystals. Phys. Rev. E 2014, 89, 012306. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, O.S.; van den Ende, D.; Stuart, M.C.; Mugele, F.; Duits, M.H.G. Hard and soft colloids at fluid interfaces: Adsorption, interactions, assembly & rheology. Adv. Colloid Interface Sci. 2015, 222, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Dhont, J.K.G. An Introduction to Dynamics of Colloids; Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Bähler, P.T.; Zanini, M.; Morgese, G.; Benetti, E.M.; Isa, L. Immobilization of Colloidal Monolayers at Fluid–Fluid Interfaces. Gels 2016, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Cristofolini, L.; Orsi, D.; Isa, L. Characterization of the dynamics of interfaces and of interface-dominated systems via spectroscopy and microscopy techniques. Curr. Opin. Colloid Interface Sci. 2018, 37, 13–32. [Google Scholar] [CrossRef]
- Boniello, G.; Blanc, C.; Fedorenko, D.; Medfai, M.; Mbarek, N.B.; In, M.; Gross, M.; Stocco, A.; Nobili, M. Brownian diffusion of a partially wetted colloid. Nat. Mat. 2015, 14, 908–911. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, W.; Fischer, T.M.; Weitz, D.A.; Tong, P. Short-time self-diffusion of nearly hard spheres at an oil-water interface. J. Fluid Mech. 2009, 618, 243–261. [Google Scholar] [CrossRef]
- Cipelletti, L.; Bissig, H.; Trappe, V.; Ballesta, P.; Mazoyer, S. Time-resolved correlation: A new tool for studying temporally heterogeneous dynamics. J. Phys. Condens. Matter 2002, 15, S257–S262. [Google Scholar] [CrossRef]
- Pusey, P.N.; Van Megen, W. Dynamic light scattering by non-ergodic media. Physica A 1989, 157, 705–741. [Google Scholar] [CrossRef]
- Mazoyer, S.; Ebert, F.; Maret, G.; Keim, P. Correlation between dynamical heterogeneities, structure and potential-energy distribution in a 2D amorphous solid. Eur. Phys. J. E 2011, 34, 101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellour, M.; Skouri, M.; Munch, J.P.; Hébraud, P. Brownian motion of particles embedded in a solution of giant micelles. Eur. Phys. J. E 2002, 8, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Keim, P.; Maret, G.; Herz, U.; von Grünberg, H.H. Harmonic Lattice Behavior of Two-Dimensional Colloidal Crystals. Phys. Rev. Lett. 2004, 92, 215504. [Google Scholar] [CrossRef] [PubMed]
- Liggieri, L.; Miller, R. (Eds.) Interfacial Rheology; Brill: Leiden, The Netherlands, 2010. [Google Scholar]
- Tajuelo, J.; Guzmán, E.; Ortega, F.; Rubio, R.G.; Rubio, M.A. Phase Diagram of Fatty Acid Langmuir Monolayers from Rheological Measurements. Langmuir 2017, 33, 4280–4290. [Google Scholar] [CrossRef] [PubMed]
- Llamas, S.; Guzmán, E.; Akanno, A.; Fernández-Peña, L.; Ortega, F.; Campbell, R.A.; Miller, R.; Rubio, R.G. Study of the Liquid/Vapor Interfacial Properties of Concentrated Polyelectrolyte–Surfactant Mixtures Using Surface Tensiometry and Neutron Reflectometry: Equilibrium, Adsorption Kinetics, and Dilational Rheology. J. Phys. Chem. C 2018, 122, 4419–4427. [Google Scholar] [CrossRef]
- Guzmán, E.; Fernández-Peña, L.; Akanno, A.; Llamas, S.; Ortega, F.; G Rubio, R. Two Different Scenarios for the Equilibration of Polycation—Anionic Solutions at Water–Vapor Interfaces. Coatings 2019, 9, 438. [Google Scholar] [CrossRef]
- Guzman, E.; Santini, E.; Ferrari, M.; Liggieri, L.; Ravera, F. Effect of the Incorporation of Nanosized Titanium Dioxide on the Interfacial Properties of 1,2-Dipalmitoyl-sn-glycerol-3-phosphocholine Langmuir Monolayers. Langmuir 2017, 33, 10715–10725. [Google Scholar] [CrossRef]
- Guzman, E.; Santini, E.; Zabiegaj, D.; Ferrari, M.; Liggieri, L.; Ravera, F. Interaction of Carbon Black Particles and Dipalmitoylphosphatidylcholine at the Water/Air Interface: Thermodynamics and Rheology. J. Phys. Chem. C 2015, 119, 26937–26947. [Google Scholar] [CrossRef]
- Guzman, E.; Ferrari, M.; Santini, E.; Liggieri, L.; Ravera, F. Effect of silica nanoparticles on the interfacial properties of a canonical lipid mixture. Colloids Surf. B 2015, 136, 971–980. [Google Scholar] [CrossRef]
- Guzman, E.; Santini, E.; Ferrari, M.; Liggieri, L.; Ravera, F. Interfacial Properties of Mixed DPPC-Hydrophobic Fumed Silica Nanoparticle Layers. J. Phys. Chem. C 2015, 119, 21024–21034. [Google Scholar] [CrossRef]
- Guzman, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. Mixed DPPC-cholesterol Langmuir monolayers in presence of hydrophilic silica nanoparticles. Colloids Surf. B 2013, 105, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. DPPC-DOPC Langmuir monolayers modified by hydrophilic silica nanoparticles: Phase behaviour, structure and rheology. Colloids Surf. A 2012, 413, 174–183. [Google Scholar] [CrossRef]
- Guzman, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. Influence of silica nanoparticles on phase behavior and structural properties of DPPC-Palmitic acid Langmuir monolayers. Colloids Surf. A 2012, 413, 280–287. [Google Scholar] [CrossRef]
- Guzman, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. Influence of silica nanoparticles on dilational rheology of DPPC-palmitic acid Langmuir monolayers. Soft Matter 2012, 8, 3938–3948. [Google Scholar] [CrossRef]
- Guzman, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. Effect of Hydrophilic and Hydrophobic Nanoparticles on the Surface Pressure Response of DPPC Monolayers. J. Phys. Chem. C 2011, 115, 21715–21722. [Google Scholar] [CrossRef]
- Sanchez-Arribas, N.; Guzman, E.; Lucia, A.; Toloza, A.C.; Velarde, M.G.; Ortega, F.; Rubio, R.G. Environmentally friendly platforms for encapsulation of an essential oil: Fabrication, characterization and application in pests control. Colloids Surf. A 2018, 555, 473–481. [Google Scholar] [CrossRef]
- Ryazantsev, Y.S.; Velarde, M.G.; Guzman, E.; Rubio, R.G.; Ortega, F.; Montoya, J.-J. On the autonomous motion of active drops or bubbles. J. Colloid Interface Sci. 2018, 527, 180–186. [Google Scholar] [CrossRef]
- Ryazantsev, Y.S.; Velarde, M.G.; Rubio, R.G.; Guzman, E.; Ortega, F.; Lopez, P. Thermo- and soluto-capillarity: Passive and active drops. Adv. Colloid Interface Sci. 2017, 247, 52–80. [Google Scholar] [CrossRef]
- Noskov, B.A. Fast adsorption at the liquid-gas interface. Adv. Colloid Interface Sci. 1996, 69, 63–129. [Google Scholar] [CrossRef]
- Bykov, A.G.; Guzmán, E.; Rubio, R.G.; Krycki, M.M.; Milyaeva, O.Y.; Noskov, B.A. Influence of temperature on dynamic surface properties of spread DPPC monolayers in a broad range of surface pressures. Chem. Phys. Lipids 2019, 225, 104812. [Google Scholar] [CrossRef]
- Ravera, F.; Santini, E.; Loglio, G.; Ferrari, M.; Liggieri, L. Effect of Nanoparticles on the Interfacial Properties of Liquid/Liquid and Liquid/Air Surface Layers. J. Phys. Chem. B 2006, 110, 19543–19551. [Google Scholar] [CrossRef]
- Ravera, F.; Ferrari, M.; Liggieri, L.; Loglio, G.; Santini, E.; Zanobini, A. Liquid–liquid interfacial properties of mixed nanoparticle–surfactant systems. Colloids Surf. A 2008, 323, 99–108. [Google Scholar] [CrossRef]
- Kotsmar, C.; Pradines, V.; Alahverdjieva, V.S.; Aksenenko, E.V.; Fainerman, V.B.; Kovalchuk, V.I.; Krägel, J.; Leser, M.E.; Noskov, B.A.; Miller, R. Thermodynamics, adsorption kinetics and rheology of mixed protein–surfactant interfacial layers. Adv. Colloid Interface Sci. 2009, 150, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Safouane, M.; Langevin, D.; Binks, B.P. Effect of Particle Hydrophobicity on the Properties of Silica Particle Layers at the Air−Water Interface. Langmuir 2007, 23, 11546–11553. [Google Scholar] [CrossRef] [PubMed]
- Zahn, K.; Wille, A.; Maret, G.; Sengupta, S.; Nielaba, P. Elastic Properties of 2D Colloidal Crystals from Video Microscopy. Phys. Rev. Lett. 2003, 90, 155506. [Google Scholar] [CrossRef]
- Zang, D.Y.; Rio, E.; Langevin, D.; Wei, B.; Binks, B.P. Viscoelastic properties of silica nanoparticle monolayers at the air-water interface. Eur. Phys. J. E 2010, 31, 125–134. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kawaguchi, M. Surface dilational moduli of latex-particle monolayers spread at air–water interface. J. Colloid Interface Sci. 2013, 390, 147–150. [Google Scholar] [CrossRef]
- del Río, O.I.; Kwok, D.Y.; Wu, R.; Alvarez, J.M.; Neumann, A.W. Contact angle measurements by axisymmetric drop shape analysis and an automated polynomial fit program1This paper represents, in part, the PhD theses of D.Y. Kwok and O.I. del Río.1. Colloids Surf. A 1998, 143, 197–210. [Google Scholar] [CrossRef]
- Cicuta, P.; Stancik, E.J.; Fuller, G.G. Shearing or Compressing a Soft Glass in 2D: Time-Concentration Superposition. Phys. Rev. Lett. 2003, 90, 236101. [Google Scholar] [CrossRef]
- Trappe, V.; Weitz, D.A. Scaling of the Viscoelasticity of Weakly Attractive Particles. Phys. Rev. Lett. 2000, 85, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, S.; Moldenaers, P.; Vermant, J. Interfacial rheology of stable and weakly aggregated two-dimensional suspensions. Phys. Chem. Chem. Phys. 2007, 9, 6463–6475. [Google Scholar] [CrossRef] [PubMed]
- Imperiali, L.; Liao, K.-H.; Clasen, C.; Fransaer, J.; Macosko, C.W.; Vermant, J. Interfacial Rheology and Structure of Tiled Graphene Oxide Sheets. Langmuir 2012, 28, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Christopher, G.F. Simultaneous Interfacial Rheology and Microstructure Measurement of Densely Aggregated Particle Laden Interfaces Using a Modified Double Wall Ring Interfacial Rheometer. Langmuir 2014, 30, 9752–9760. [Google Scholar] [CrossRef] [PubMed]
- Madivala, B.; Fransaer, J.; Vermant, J. Self-Assembly and Rheology of Ellipsoidal Particles at Interfaces. Langmuir 2009, 25, 2718–2728. [Google Scholar] [CrossRef]
- Brown, E.; Zhang, H.; Forman, N.A.; Maynor, B.W.; Betts, D.E.; DeSimone, J.M.; Jaeger, H.M. Shear thickening and jamming in densely packed suspensions of different particle shapes. Phys. Rev. E 2011, 84, 031408. [Google Scholar] [CrossRef]
- Wilson, H.J.; Davis, R.H. Shear stress of a monolayer of rough spheres. J. Fluid Mech. 2002, 452, 425–441. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).