1. Introduction
Chemical absorption is widely used and the most mature technology to remove CO
2 from gas streams. However, it is known that the energy consumption to regenerate the solvent is one of the biggest concerns of this type of process. Other major concerns include solvent emissions, stability, and equipment size. Several studies on solvent development (e.g., phase change solvent [
1], new blends [
2,
3] and water lean solvents [
4]) and on process modification [
5,
6] aiming to address these issues have been done since Bottoms patented in 1930 a process to remove acid gases from natural gas.
To design and simulate effectively an absorption process, information is required on both the solvent properties (e.g., physical properties, vapor liquid equilibrium, and absorption kinetics) and the equipment (e.g., absorber type and packing material). Traditionally, in the absorption process, the absorption and desorption are performed in columns: the absorber and the stripper columns, respectively. However, some new designs have been recently proposed. For example, Lin et al. (2016) [
7] studied the desorption step using the advanced flash stripper configuration for a 5 m and a 8 m piperazine (PZ) solution. They showed that the process was more energy efficient than the benchmark solvent monoethanolamine (MEA) and previous process configuration, namely the two-staged flash. A reboiler duty of 2.1–2.5 GJ/ton CO
2 was achieved. Recently, membrane contactors have been studied to substitute the absorber tower [
8]. Using a membrane contactor could potentially reduce significantly the equipment size as calculated in Hoff and Svendsen (2013) [
9].
Traditional solvents such as monoethanolamine (MEA), methyldiethanolamine (MDEA), and piperazine (PZ) have been widely studied and their behavior is well represented by several commercial softwares. However, new solvents/solvent blends are being developed to improve the efficiency and safety of the absorption process. In the solvent development the vapor-liquid equilibria, heat of absorption, corrosion tendencies, solvent degradation, absorption kinetics, and solvent volatility are among the properties that are experimentally measured. In recent years, solvent volatility has gained a lot of attention. Volatility causes solvent losses requiring water wash sections to control the emissions. Volatility in combination with mist formation can significantly magnify the solvent losses. There are three strategies to overcome the volatility issue. One is to develop solvents with very low volatility, like aqueous amino acid salt solutions [
10]. The second is to develop systems/operations that minimize the formation of mist, like the anti-mist design developed by Aker Solutions [
11]. In recent years, a third strategy has been proposed: the use of a non-porous thin composite membrane [
8]. This type of membrane can potentially reduce the amine evaporation towards the gas phase.
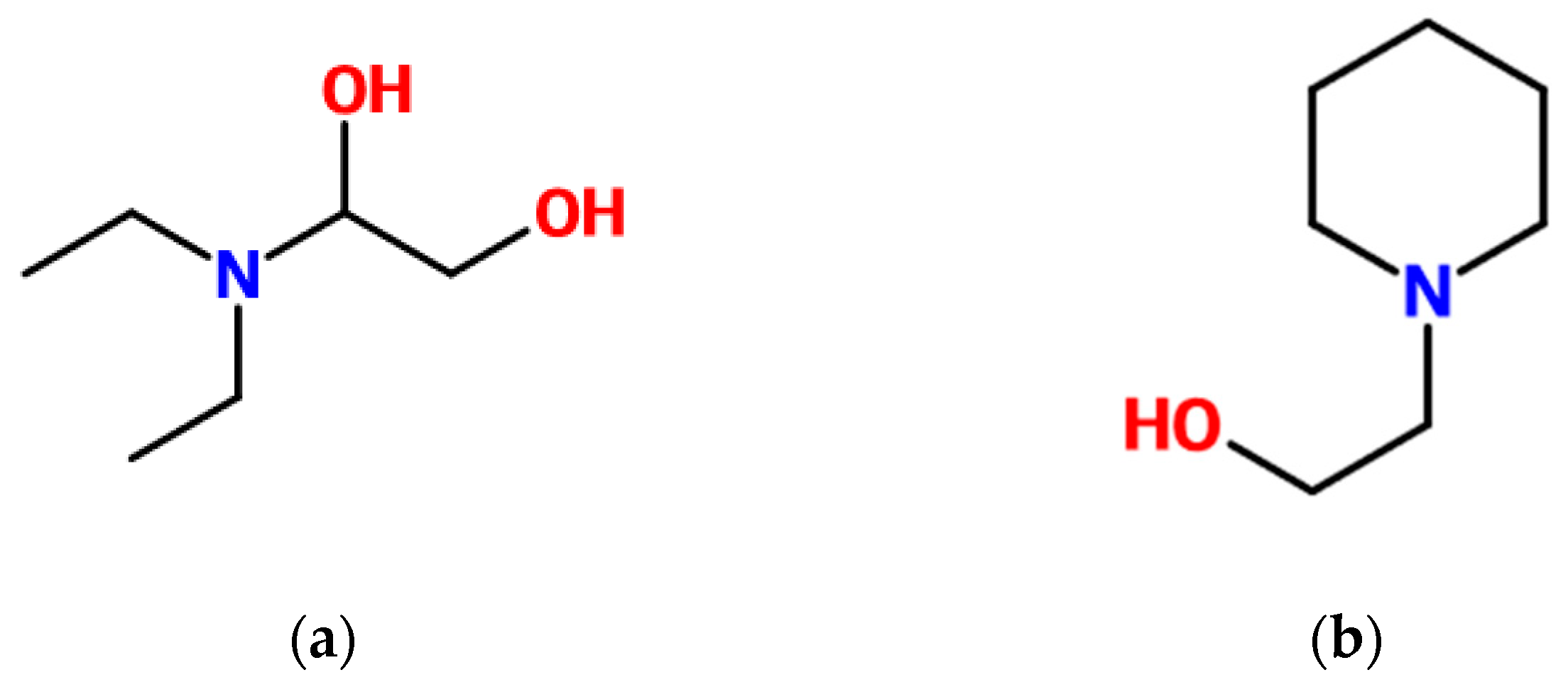
Independently of the equipment used for the absorption process, a good description of the vapor liquid equilibrium, together with other properties, is essential. 3-(Diethylamino)-1,2-propanediol (DEA-12-PD) and 1-(2-hydroxyethyl)piperidine (12-HEPP) have been proposed as potential components in solvent blends for membrane contactors [
12]. Very few experimental data are found for these two tertiary amines. DEA-12-PD had been identified by Chowdhury et al. (2013) [
13] as a potential tertiary amine since the absorption rate and capacity were good. Li et al. (2015) [
14] studied the reaction kinetics of aqueous solutions of DEA-12-PD with CO
2 and it was observed that the reaction was faster than MDEA. They also performed pKa measurements at different temperatures.
Later Hartono et al. (2017) [
15] performed a series of screening tests with different solvents that could intensify the formation of bicarbonate. DEA-12-PD and 12-HEPP were among the tested solvent candidates. Knuutila et al. (2019) [
3] tested DEA-12-PD and 12-HEPP promoted with primary amines and showed that by using a short cut method [
16], the tested solvent blends could be regenerated with reboiler duties 2.5–2.6 MJ/kg CO
2. These values are similar to those values measured with several novel solvent blends, 2.5–3.0 MJ/kg CO
2 [
17,
18,
19].
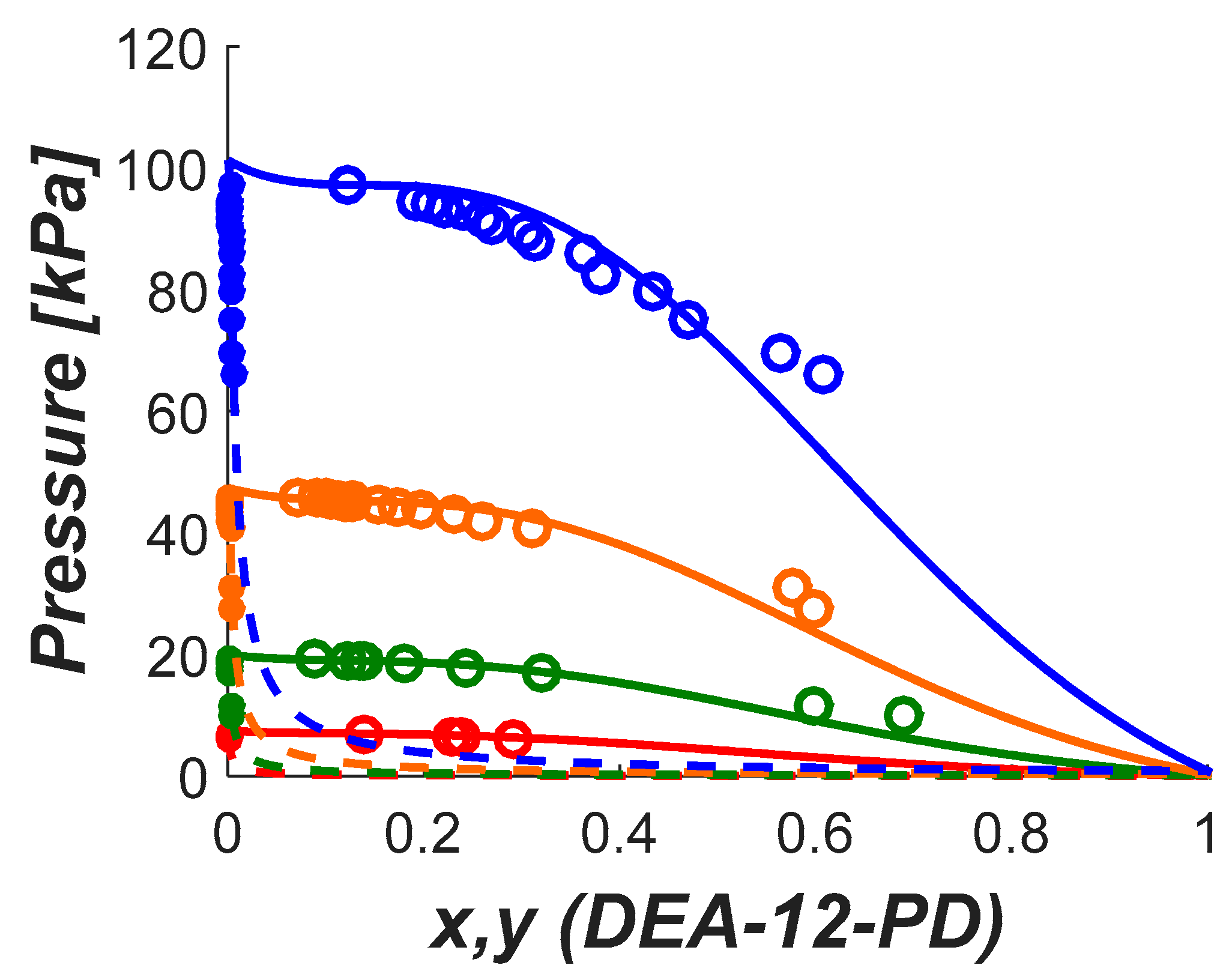
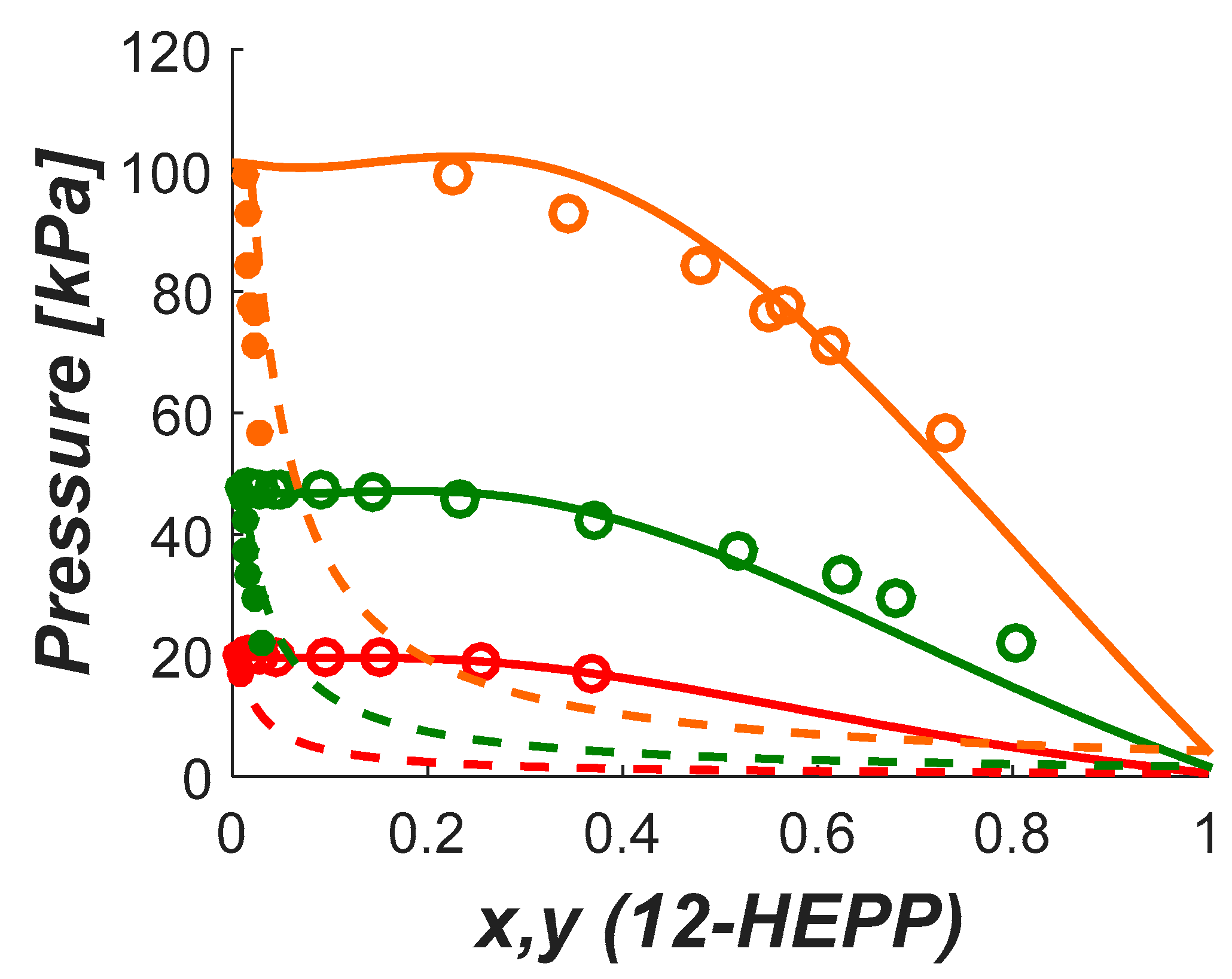
No data was found for the vapor liquid equilibrium of aqueous DEA-12-PD and 12-HEPP systems. Understanding the volatility of solvent components is an important parameter as discussed above. The volatility will be influenced by the CO2 loading and the degree of solvent degradation, both depending on the actual industrial application. However, a good estimation of the potential challenges related to volatility can be gained by measuring the vapor–liquid equilibria of non-degraded binary systems.
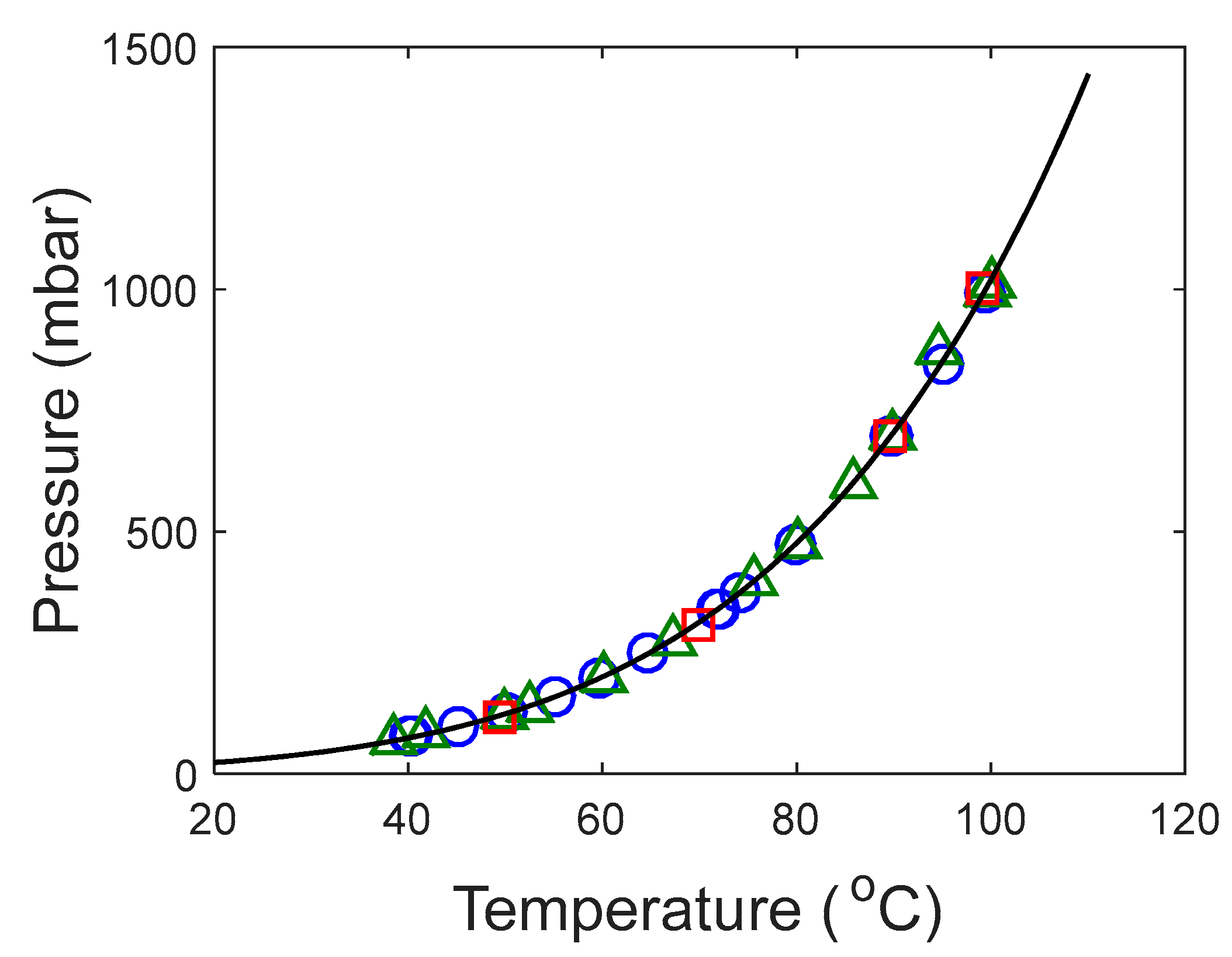
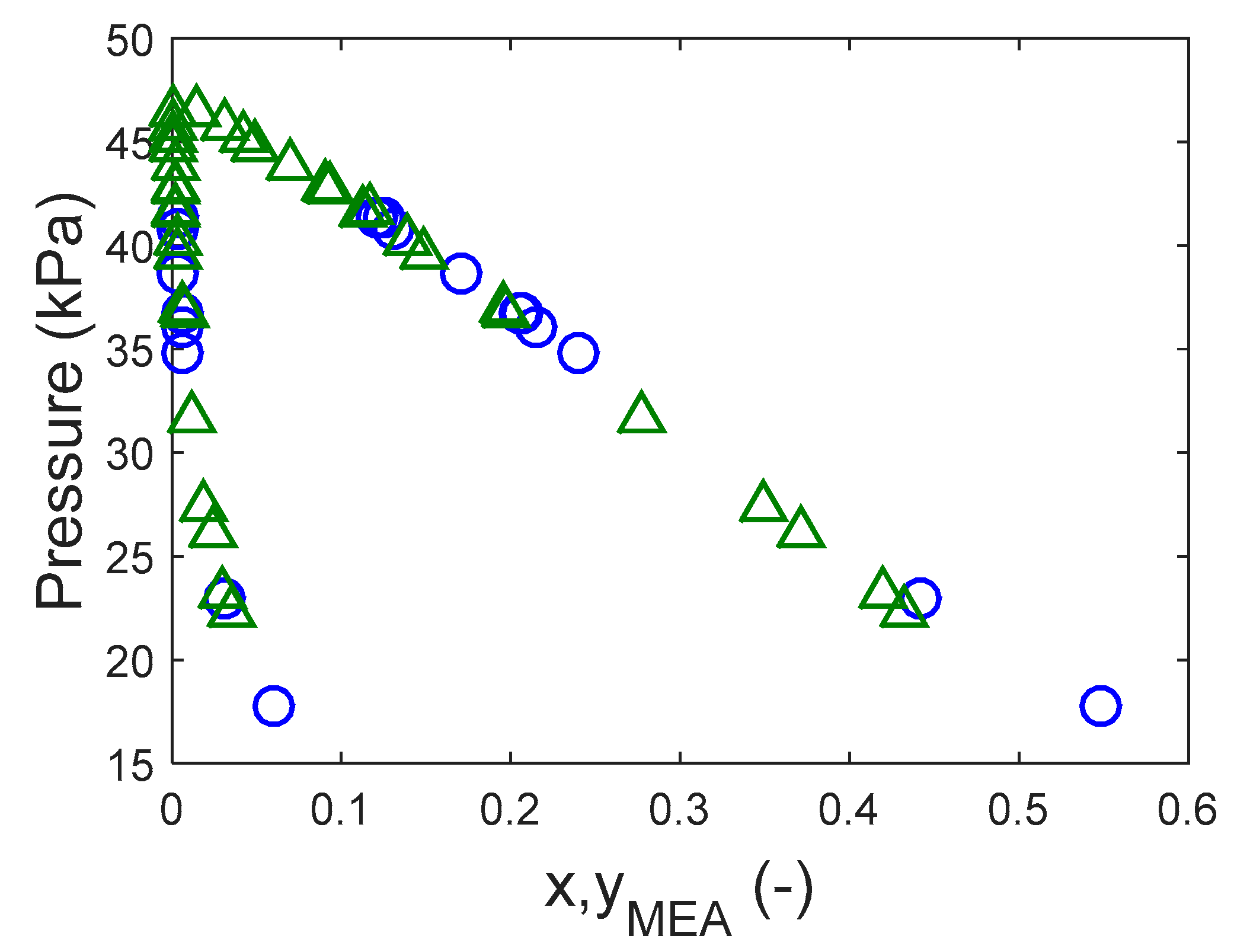
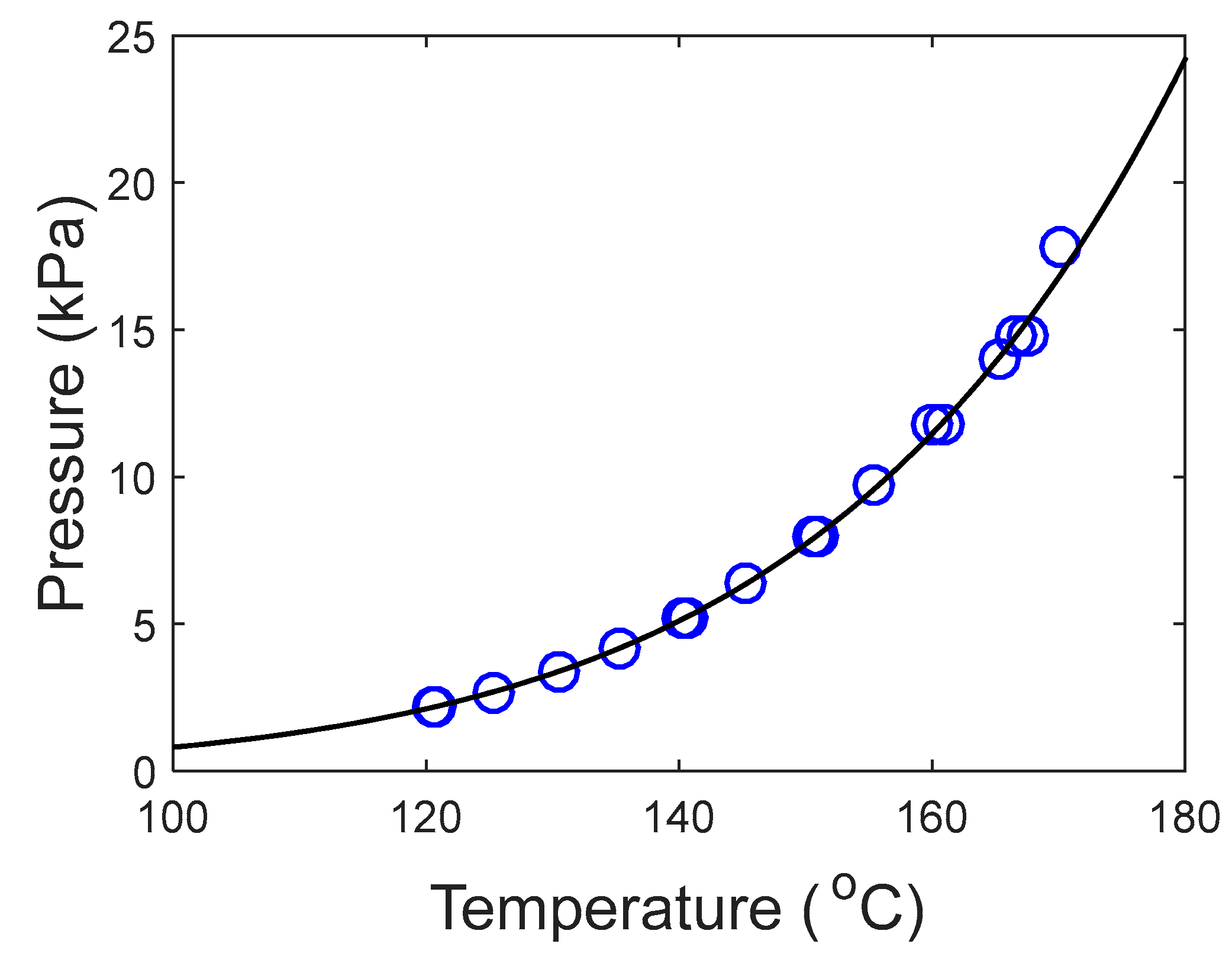
In this work, we provide experimental data on the pure component saturation pressure and on VLE of aqueous solutions of these amines. The data were then used to estimate model parameters used to represent the data. The saturation pressure was modeled using the Antoine equation and the deviation was calculated lower than 2%. The NRTL model was used in this work to calculate the activity coefficients in the aqueous systems. The deviations in pressure for the aqueous systems were lower than 5% in both systems.
3. Modeling
The Antoine equation, Equation (1), was used to correlate the saturation pressure of both pure DEA-12-PD and 12-HEPP. This correlation is frequently used to represent the saturation pressure of pure components and, in this work, it was chosen to represent the produced data.
In this work, a non-electrolyte non-reactive system was assumed for the representation of the VLE of aqueous DEA-12-PD and 12-HEPP. The phase equilibrium is solved iteratively for the pressure and vapor phase composition by solving the system of equations described in Equation (2). In that equation the subscript stands for H2O and the amine, is the activity coefficient, is the fugacity coefficient, and are the liquid and gas phase mol fractions, respectively, and the exponential term is the so-called Poynting correction factor where the liquid volume was fixed as the respective component molar volume. As expected, since the experiments were carried out under low pressures, the Poynting correction factor for the conditions studied in this work was negligible.
The Peng–Robinson equation of state (EoS) [
24] is used to correct the gas phase non-idealities while the non-random two-liquids (NRTL) model [
25] is used to account for the liquid phase non-ideal behavior. The Van der Waals mixing rule with all binary parameters set to zeros was used in the Peng–Robinson EoS. As a result, all adjustable parameters are from the NRTL model.
The activity coefficient calculated through the NRTL model is given in Equation (3). The binary energy parameters are assumed to have a temperature dependency as shown in Equation (5) where
and
are adjustable parameters. The non-randomness parameter
can also be used as an adjustable parameter, but in this work it was fixed at a given value.
Besides the rigorous framework, the choice of the set of models used in this work, among others, was based on the ease of exporting the parameters to process simulation software. Most process simulators have the models used in this work already implemented.
3.1. Optimization Routine
The adjustable parameters from the NRTL model were fitted to the experimental data using the particle swarm optimization (PSO) routine with the local best topology. The method is widely used for parameter estimation [
23,
26,
27,
28] and information about it can be found elsewhere (e.g., [
23,
29]). In this work, we fixed the non-randomness parameters at 0.1, 0.2, and 0.3 using four different objective functions. The best results are given in the results section while the results from all optimizations can be found in
Appendix B.
Equations (6) and (7) show the general form of the objective functions used in this work. In those equations, the parameter q was set to zero if the vapor phase composition was not included in the objective function. In the case that the vapor phase composition should be considered in the optimization, the parameter was set to one. A total of four objective functions were used per non-randomness parameter, giving a total of 12 optimizations and a set of parameters for each binary system.
The results were evaluated by means of the average absolute relative deviation (AARD) function (Equation (8)) where
is the variable from which the deviation is calculated.
3.2. Critical Properties
Since the Peng–Robinson EoS was used, the critical properties of the components were required. These properties were not found for the tertiary amines studied in this work. Therefore, the Joback group contribution method [
30] was used for this purpose. For 12-HEPP, the tertiary amine contribution was considered as a “non-ring” tertiary amine contribution since the method has no “ring” tertiary amine contribution. Nevertheless, the estimations given here should be treated with care as these results must be experimentally confirmed. The normal boiling point for the amines was calculated with the Antoine equation fitted with the respective experimental points generated in this work.
As a comparison, the critical properties for MEA were estimated using the Joback group contribution method. Using a normal boiling temperature of 443.97 K, the critical temperature, pressure, and volume were estimated to be 637.1 K, 62.89 bar, and 1.96 m
3/mol, respectively. The reported values for the critical temperature, pressure, and volume for MEA [
31] are, respectively, 678.2 K, 71.24 bar, and 2.25 m
3/mol. This shows that using the Joback group contribution method gives a good initial estimation for the critical point of a substance.
Once the critical properties are known, the acentric factor can be calculated using Equation (9). The results are summarized in
Table 2.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






