A Glucose-Dependent Pharmacokinetic/ Pharmacodynamic Model of ACE Inhibition in Kidney Cells



Abstract
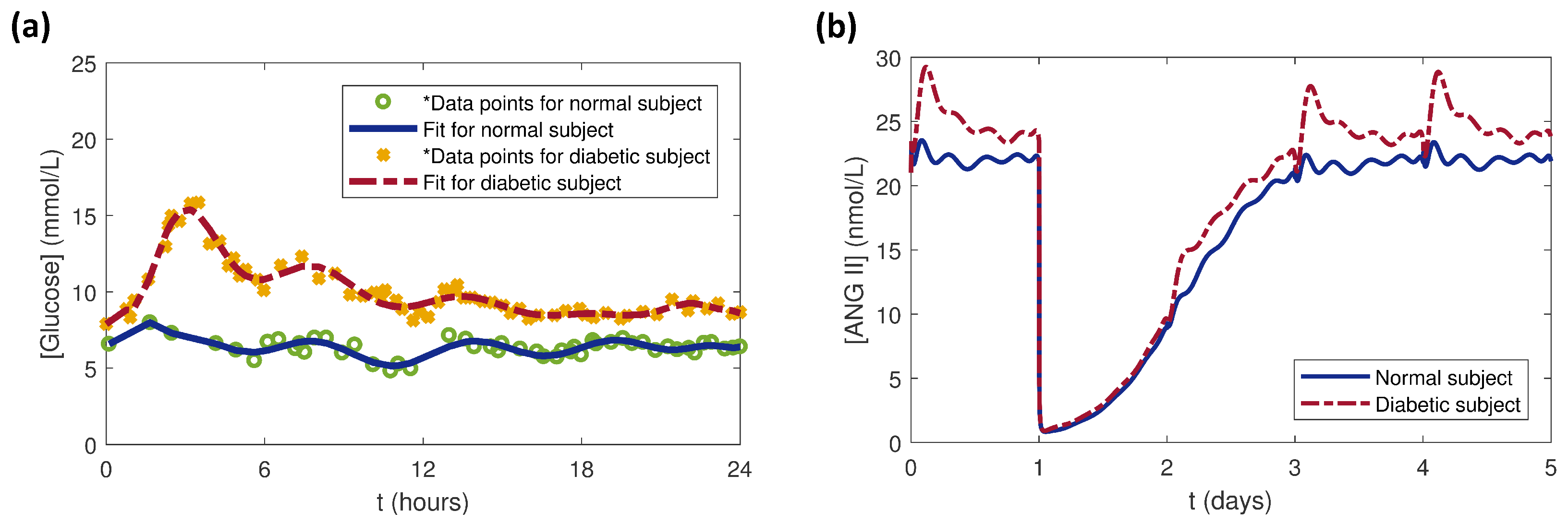
:1. Introduction
2. Methods
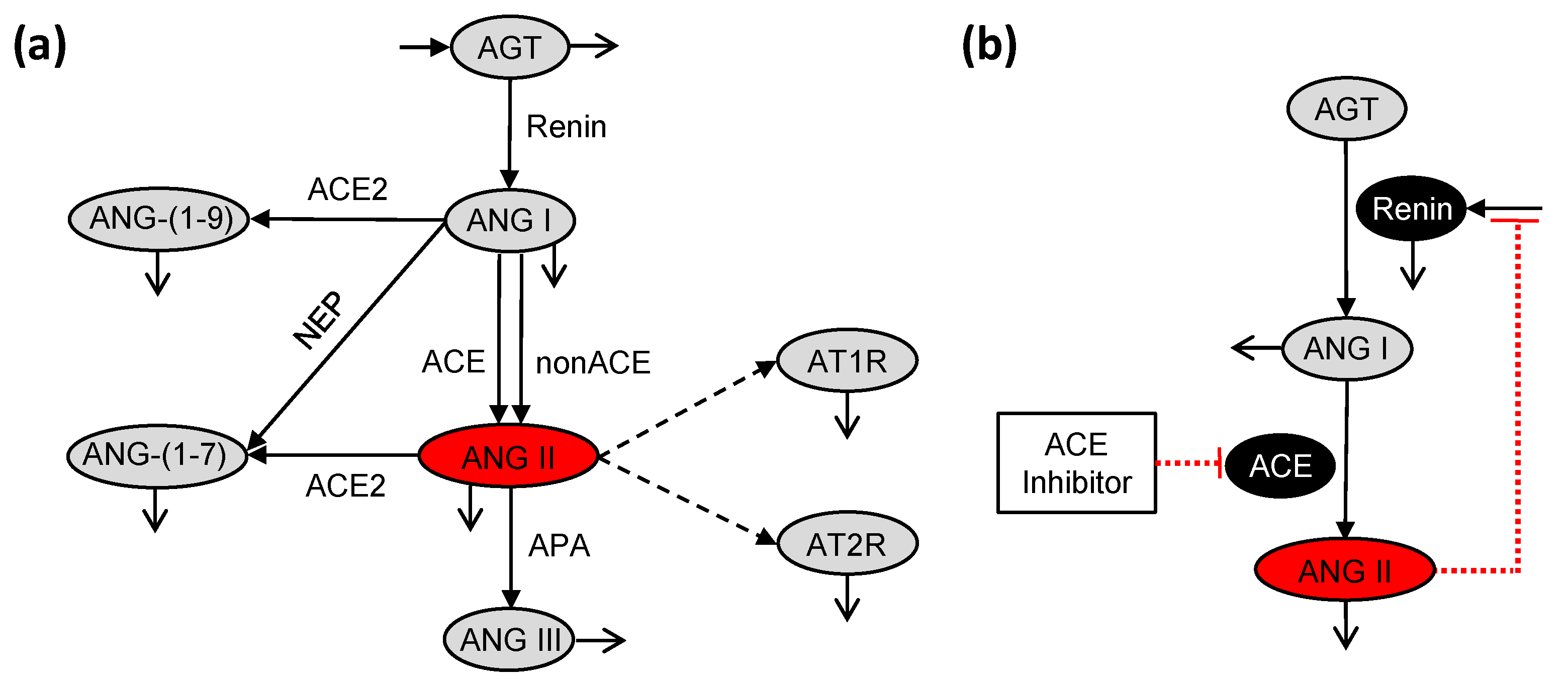
2.1. Pharmacokinetic Model
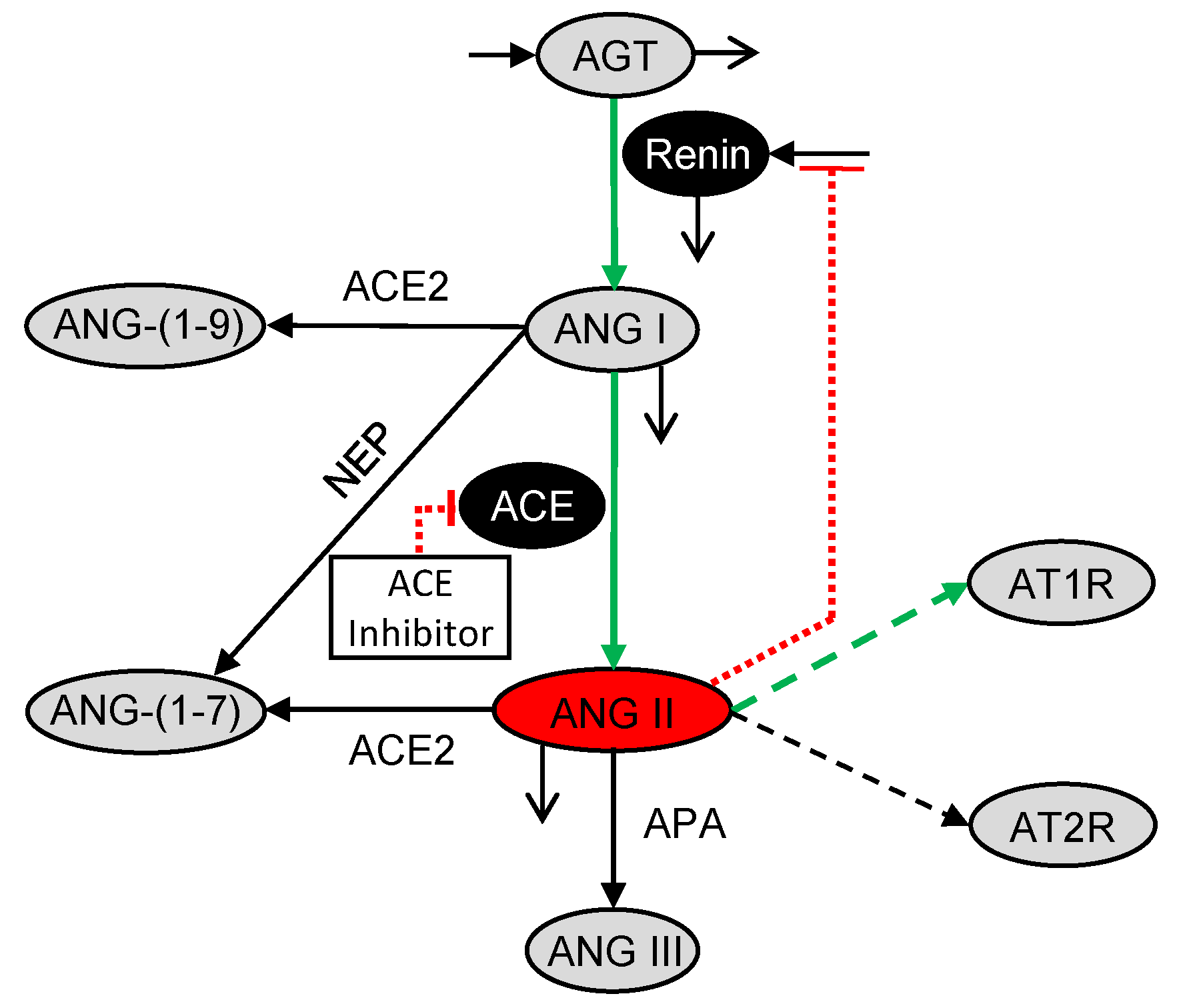
2.2. Pharmacodynamic Model
2.3. Numerical Methods and Code Repository
3. Results and Discussion
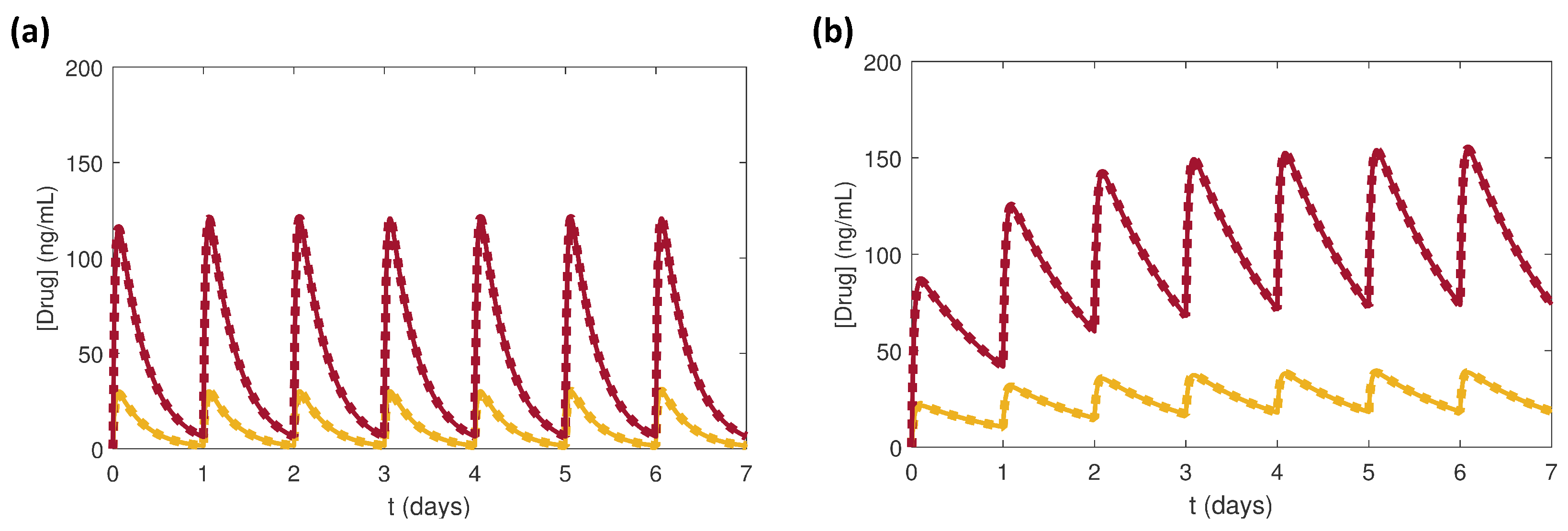
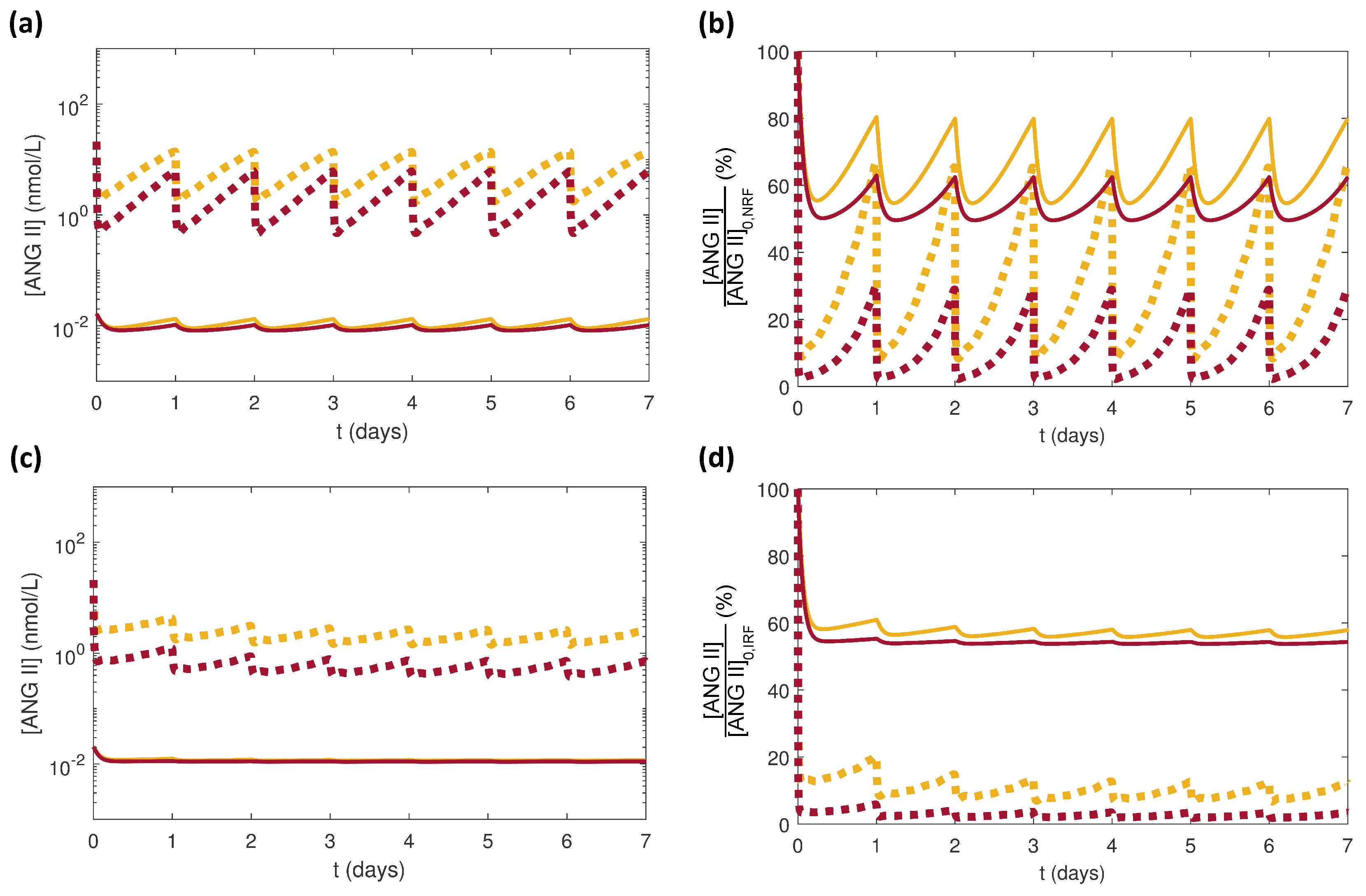
3.1. Effects of Doses of Benazepril
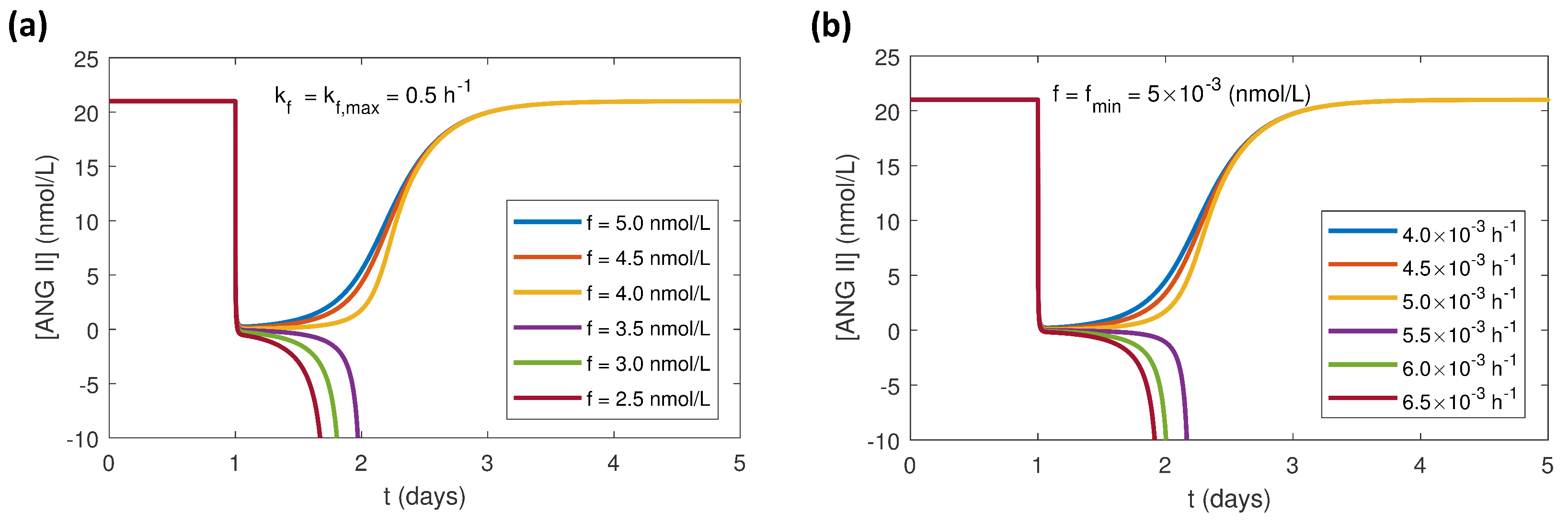
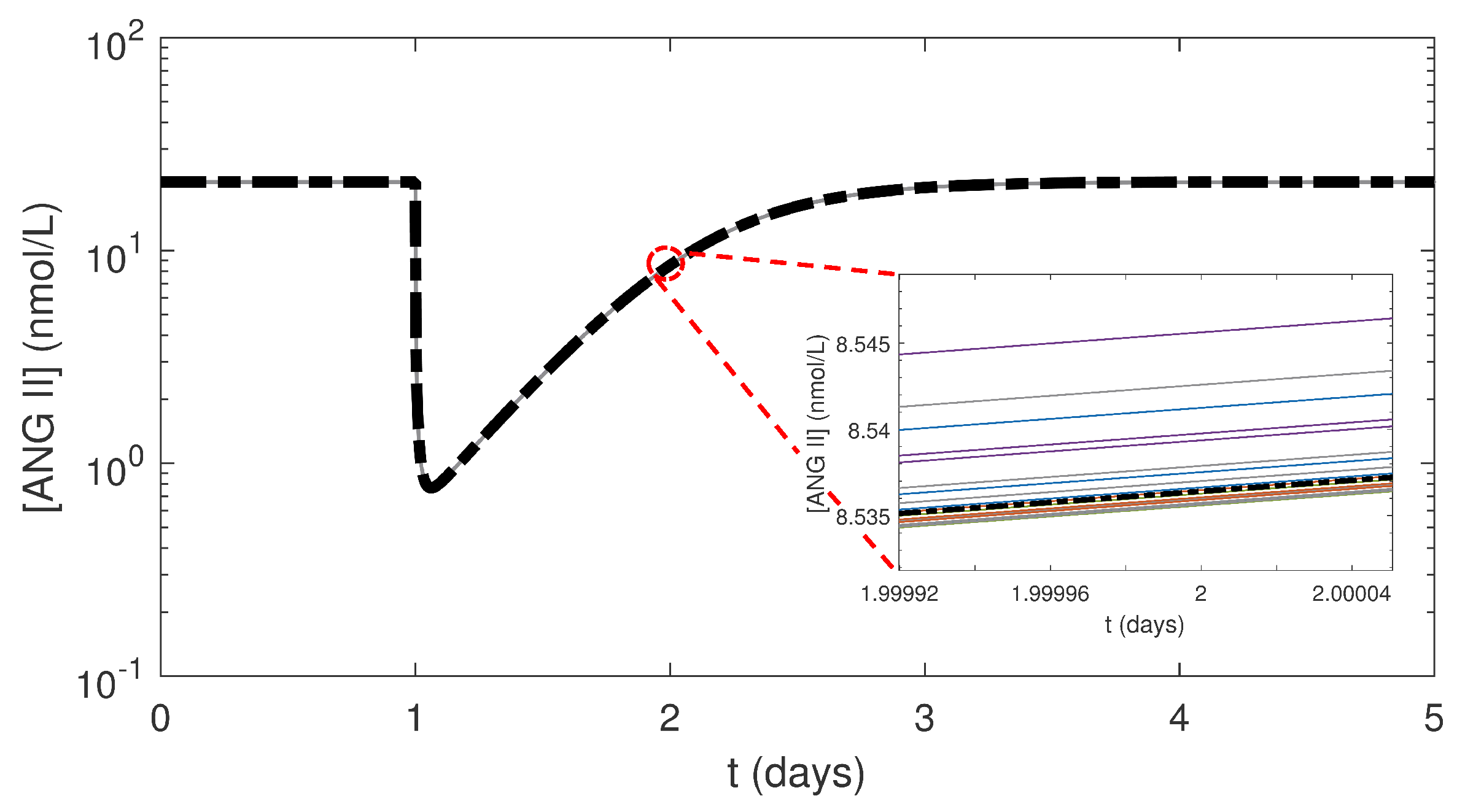
3.2. Effects of Varying the Uncertain Feedback Parameters
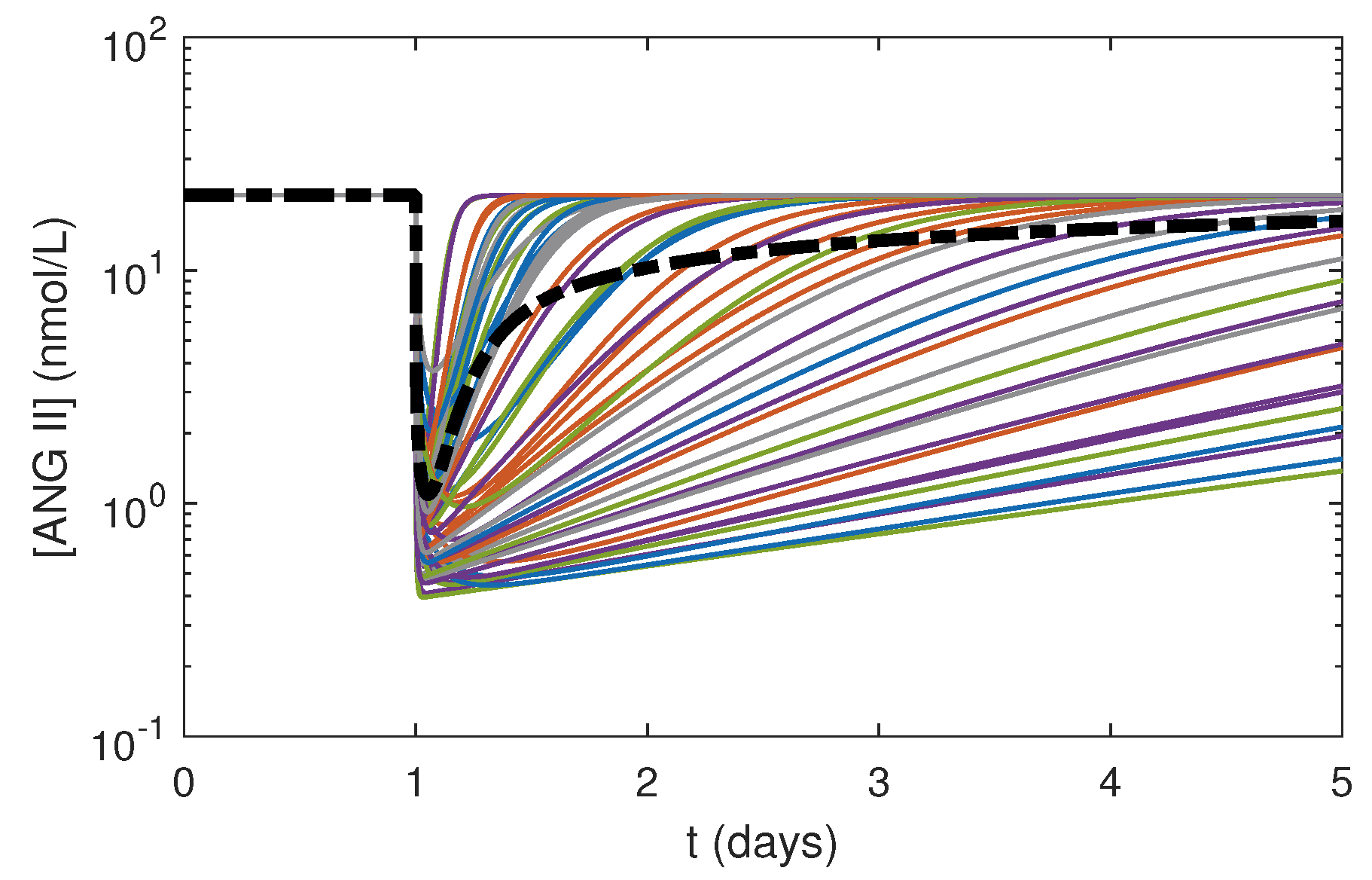
3.3. Effects of Varying PK Parameters
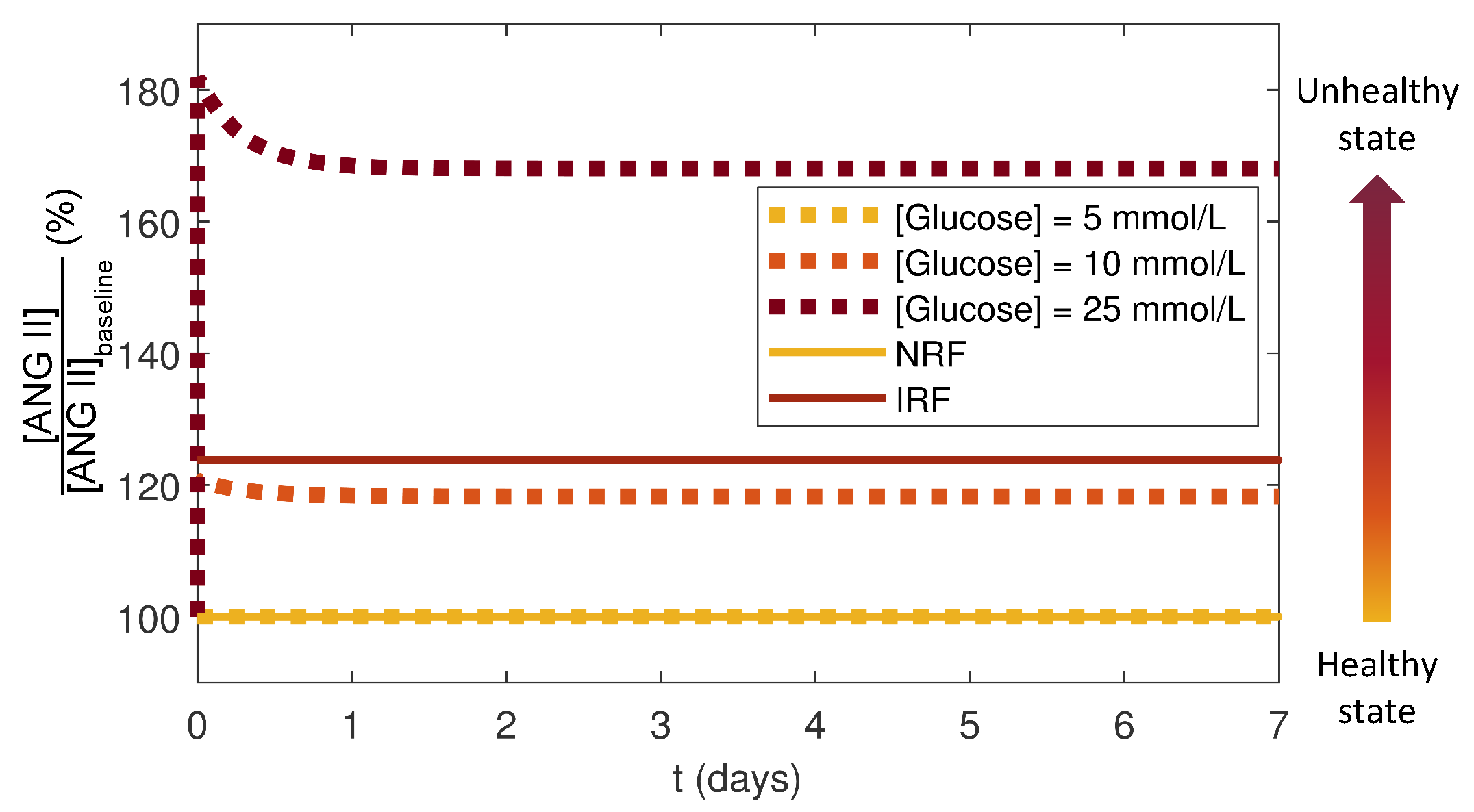
3.4. Effects of Glucose Conditions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garg, P.; Holzman, L.B. Podocytes: Gaining a foothold. Exp. Cell Res. 2012, 318, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Martinez-Salgado, C.; Rodriguez-Pena, A.B.; Lopez-Hernandez, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.S.; Susztak, K. Podocytes: The weakest link in diabetic kidney disease? Curr. Diab. Rep. 2016, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Levchenko, V.; Lowing, A.; Shuyskiy, L.S.; Palygin, O.; Staruschenko, A. Podocyte injury in diabetic nephropathy: Implications of angiotensin II-dependent activation of TRPC channels. Sci. Rep. 2015, 5, 17637. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Liu, Q.; Liu, B. Research progress on mechanism of podocyte depletion in diabetic nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.S. Glomerular endothelial cells stress and cross-talk with podocytes in the development of diabetic kidney disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Márquez, E.; Riera, M.; Pascual, J.; Soler, M.J. Renin-angiotensin system within the diabetic podocyte. Am. J. Physiol. Ren. Physiol. 2015, 308, F1–F10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danser, A.H.J.; Admiraal, P.J.J.; Derkx, F.H.M.; Schalekamp, M.A.D.H. Angiotensin I-to-II conversion in the human renal vascular bed. J. Hypertens. 1998, 16, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Liebau, M.C.; Lang, D.; Böhm, J.; Endlich, N.; Bek, M.J.; Witherden, I.; Mathieson, P.W.; Saleem, M.A.; Pavenstädt, H.; Fischer, K.G. Functional expression of the renin-angiotensin system in human podocytes. Am. J. Physiol. Ren. Physiol. 2006, 290, F710–F719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez, J.C.Q.; Bland, A.M.; Arthur, J.M.; Raymond, J.R.; Janech, M.G. Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am. J. Physiol. Ren. Physiol. 2007, 293, F398–F407. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Shankland, S.J. Activation of a local renin angiotensin system in podocytes by glucose. Am. J. Physiol. Ren. Physiol. 2008, 294, F830–F839. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.H.; Li, J.J.; Kim, J.J.; Jung, D.S.; Kwak, S.J.; Ryu, D.R.; Choi, H.Y.; Kim, J.S.; Kim, H.J.; Han, S.H.; et al. Activation of the renin-angiotensin system within podocytes in diabetes. Kidney Int. 2007, 71, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Seth, D.M.; Navar, L.G. Renal interstitial fluid angiotensin I and angiotensin II concentrations during local angiotensin-converting enzyme inhibition. J. Am. Soc. Nephrol. 2002, 13, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Beh, J.; De Leon, H.; Hallow, M.K.; Ramakrishna, R.; Rodrigo, M.; Sarkar, A.; Sarangapani, R.; Georgieva, A. Using a systems biology approach to explore hypotheses underlying clinical diversity of the renin angiotensin system and the response to antihypertensive therapies. In Clinical Trial Simulations; Kimko, H.H.C., Peck, C.C., Eds.; Springer: New York, NY, USA, 2011; pp. 457–482. [Google Scholar]
- Ford Versypt, A.N.; Harrell, G.K.; McPeak, A.N. A pharmacokinetic/pharmacodynamic model of ACE inhibition of the renin-angiotensin system for normal and impaired renal function. Comp. Chem. Eng. 2017, 104, 311–322. [Google Scholar] [CrossRef]
- Leete, J.; Gurley, S.; Layton, A.T. Modeling sex differences in the renin angiotensin system and the efficacy of antihypertensive therapies. Comp. Chem. Eng. 2018, 112, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Ford Versypt, A.N. ACEInhibPKPD. 2017. Available online: http://github.com/ashleefv/ACEInhibPKPD (accessed on 16 January 2017).
- Deen, W.M.; Bohrer, M.P.; Brenner, B.M. Macromolecule transport across glomerular capillaries: Application of pore theory. Kidney Int. 1979, 16, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R. Kidney modeling and systems physiology. WIREs Syst. Biol. Med. 2009, 1, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A. Modeling transport in the kidney: Investigating function and dysfunction. Am. J. Physiol. Ren. Physiol. 2010, 298, F475–F484. [Google Scholar] [CrossRef] [PubMed]
- Layton, A.T. Mathematical modeling of kidney transport. WIREs Syst. Biol. Med. 2013, 5, 557–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, B.C.; Edwards, A.; Sgouralis, I.; Layton, A.T. Impact of renal medullary three-dimensional architecture on oxygen transport. Am. J. Physiol. Ren. Physiol. 2014, 307, F263–F272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, A.M. A mathematical model of rat proximal tubule and loop of Henle. Am. J. Physiol. Ren. Physiol. 2015, 308, F1076–F1097. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.M. A mathematical model of the rat nephron: Glucose transport. Am. J. Physiol. Ren. Physiol. 2015, 308, F1098–F1118. [Google Scholar] [CrossRef] [PubMed]
- Arciero, J.C.; Ellwein, L.; Ford Versypt, A.N.; Makrides, E.; Layton, A.T. Modeling blood flow control in the kidney. In Applications of Dynamical Systems in Biology and Medicine; Jackson, T., Radunskaya, A., Eds.; Springer: New York, NY, USA, 2015; pp. 55–74. [Google Scholar]
- Ford Versypt, A.N.; Makrides, E.; Arciero, J.C.; Ellwein, L.; Layton, A.T. Bifurcation study of blood flow control in the kidney. Math. Biosci. 2015, 263, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgouralis, I.; Layton, A.T. Mathematical modeling of renal hemodynamics in physiology and pathophysiology. Math. Biosci. 2015, 264, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T.; Vallon, V.; Edwards, A. A mathematical model of the rat nephron: Glucose transport. Am. J. Physiol. Ren. Physiol. 2016, 310, F1269–F1283. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yi, H.; Zhao, W.; Ma, J.X.; Zhang, Y.; Zhou, X. Intraglomerular crosstalk elaborately regulates podocyte injury and repair in diabetic patients insights from a 3D multiscale modeling study. Oncotarget 2016, 7, 73130–73146. [Google Scholar] [CrossRef] [PubMed]
- Pilvankar, M.R.; Higgins, M.A.; Ford Versypt, A.N. Mathematical model for glucose dependence of the local renin-angiotensin system in podocytes. Bull. Math. Biol. 2018, 80, 880–905. [Google Scholar] [CrossRef] [PubMed]
- Pilvankar, M.R.; Higgins, M.A.; Ford Versypt, A.N. glucoseRASpodocytes. 2017. Available online: http://github.com/ashleefv/glucoseRASpodocytes (accessed on 11 June 2017).
- Mishina, M.; Watanabe, T. Development of hypertension and effects of benazepril hydrochloride in a canine remnant kidney model of chronic renal failure. J. Vet. Med. Sci. 2008, 70, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Mori, H.; Masaki, T.; Nishiyama, A. Angiotensin II blockade and renal protection. Curr. Pharm. Des. 2013, 19, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.; Sangalli, F.; Perticucci, E.; Aros, C.; Viscarra, C.; Perna, A.; Remuzzi, A.; Bertocchi, F.; Fagiani, L.; Remuzzi, G.; et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003, 63, 1094–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, I.M.; Cravedi, P.; Remuzzi, G. The role of renin angiotensin system inhibition in kidney repair. Fibrogenesis Tissue Repair 2010, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, J.P.; Sarver, J.G. Chapter 10: Pharmacokinetic modeling. In Pharmacology: Principles and Practice; Hacker, M., Bachmann, K., Messer, W., Eds.; Academic Press: New York, NY, USA, 2009; pp. 201–277. [Google Scholar]
- Shionoiri, H.; Ueda, S.I.; Minamisawa, K.; Minamisawa, M.; Takasaki, I.; Sugimoto, K.; Gotoh, E.; Ishii, M. Pharmacokinetics and pharmacodynamics of benazepril in hypertensive patients with normal and impaired renal function. J. Cardiovasc. Pharmacol. 1992, 20, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Pilvankar, M.R.; Yong, H.L.; Ford Versypt, A.N. COMBINEDglucoseRASpodocytesACEInhibPKPD. 2018. Available online: http://github.com/ashleefv/COMBINEDglucoseRASpodocytesACEInhibPKPD (accessed on 27 January 2019).
- Yard, B.; Feng, Y.; Keller, H.; Mall, C.; van der Woude, F. Influence of high glucose concentrations on the expression of glycosaminoglycans and N-deacetylase/N-sulphotransferase mRNA in cultured skin fibroblasts from diabetic patients with or without nephropathy. Nephrol. Dial. Transplant. 2002, 17, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Das, F.; Ghosh-Choudhury, N.; Dey, N.; Bera, A.; Mariappan, M.M.; Kasinath, B.S.; Choudhury, G.G. High glucose forces a positive feedback loop connecting Akt kinase and FoxO1 transcription factor to activate mTORC1 kinase for mesangial cell hypertrophy and matrix protein expression. J. Biol. Chem. 2014, 289, 32703–32716. [Google Scholar] [CrossRef] [PubMed]
- Praet, S.F.; Manders, R.J.; Meex, R.C.; Lieverse, A.; Stehouwer, C.D.; Kuipers, H.; Keizer, H.A.; Van Loon, L.J. Glycaemic instability is an underestimated problem in type II diabetes. Clin. Sci. 2006, 111, 119–126. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.D.; Beckman, R.J.; Conover, W.J. A comparison of three methods for selecting values of input variables in the analysis of output from a computer code. Technometrics 1979, 21, 239–245. [Google Scholar]
- Marino, S.; Hogue, I.B.; Ray, C.J.; Kirschner, D.E. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J. Theor. Biol. 2008, 254, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; De Vriese, A.S.; Flyvbjerg, A. From hyperglycemia to diabetic kidney disease: the role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr. Rev. 2004, 25, 971–1010. [Google Scholar] [CrossRef] [PubMed]
- SourceForge. Plot Digitizer. 2015. Available online: http://plotdigitizer.sourceforge.net/ (accessed on 2 January 2017).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NRF | IRF | Units | Sources |
|---|---|---|---|---|
| 1.907 | 1.645 | h | [15,37] | |
| 1.33 × 10 | 3.45 × 10 | h | [15,37] | |
| 7.09 × 10 | 1.07 × 10 | mL | [15,37] |
| Parameter | Value | Units | Sources |
|---|---|---|---|
| nmol/L/h | [30] | ||
| h | [15] | ||
| h | [30] | ||
| h | [30] | ||
| h | [30] | ||
| h | [30] | ||
| h | [14,30] | ||
| h | [15] | ||
| h | [14,30] | ||
| h | [14,30] | ||
| 2.20 | ng/mL | [15,37] | |
| m | 0.99 | - | [15,37] |
| L/mmol/h | [30] | ||
| h | [30] | ||
| L/mmol/h | [30] | ||
| 163 | h | [30] | |
| L/mmol/h | [30] | ||
| 464 | h | [30] | |
| h | [15] | ||
| nmol/L | [15] | ||
| nmol/L | [30] | ||
| nmol/L | [15] | ||
| 271 | nmol/L | [30] | |
| nmol/L | [30] | ||
| nmol/L | [15] | ||
| nmol/L | [15] |
| Parameter | Nominal Value | Min | Max | Units | Range |
|---|---|---|---|---|---|
| 4.91 × 10 | 5 × 10 | 5 × 10 | h | ||
| f | 5.04 × 10 | 5 × 10 | 5 × 10 | nmol/L |
| Combination | f | ||
|---|---|---|---|
| 1 | min | min | |
| 2 | min | max | |
| 3 | max | max | |
| 4 | max | min |
| Parameter | Nominal Value | Min | Max | Units | Range |
|---|---|---|---|---|---|
| 1.907 | 1.907 × 10 | 1.907 × 10 | h | ||
| 0.133 | 0.133 × 10 | 0.133 × 10 | h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilvankar, M.R.; Yong, H.L.; Ford Versypt, A.N. A Glucose-Dependent Pharmacokinetic/ Pharmacodynamic Model of ACE Inhibition in Kidney Cells. Processes 2019, 7, 131. https://doi.org/10.3390/pr7030131
Pilvankar MR, Yong HL, Ford Versypt AN. A Glucose-Dependent Pharmacokinetic/ Pharmacodynamic Model of ACE Inhibition in Kidney Cells. Processes. 2019; 7(3):131. https://doi.org/10.3390/pr7030131
Chicago/Turabian StylePilvankar, Minu R., Hui Ling Yong, and Ashlee N. Ford Versypt. 2019. "A Glucose-Dependent Pharmacokinetic/ Pharmacodynamic Model of ACE Inhibition in Kidney Cells" Processes 7, no. 3: 131. https://doi.org/10.3390/pr7030131
APA StylePilvankar, M. R., Yong, H. L., & Ford Versypt, A. N. (2019). A Glucose-Dependent Pharmacokinetic/ Pharmacodynamic Model of ACE Inhibition in Kidney Cells. Processes, 7(3), 131. https://doi.org/10.3390/pr7030131